Aicinām vēl šonedēļ piedalīties lietotāju apmierinātības aptaujā!
Paldies par viedokli!

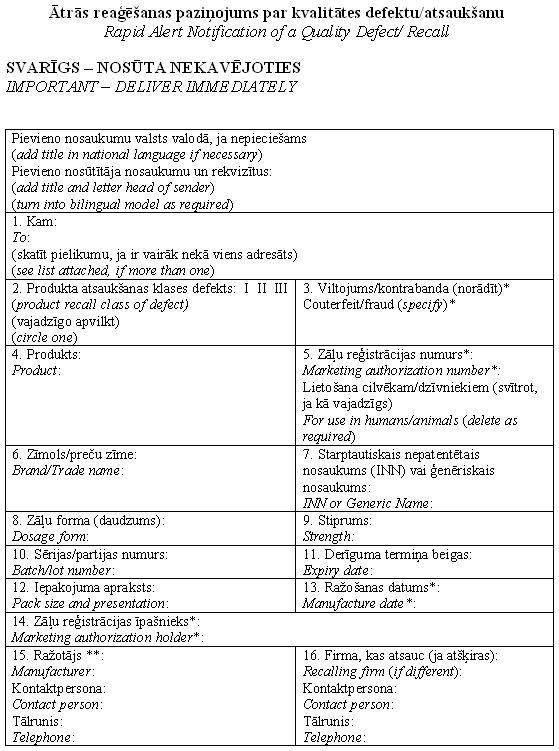
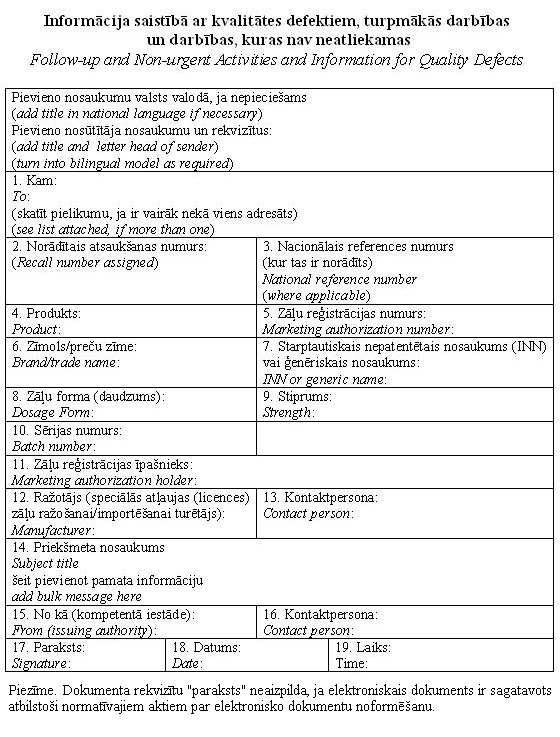
|
The translation of this document is outdated.
Translation validity: 21.12.2021.–10.07.2025. Amendments not included: 08.07.2025.
Procedures Regarding the Distribution and Quality Control of Medicinal ProductsIssued pursuant to [8 October 2013] I. General Provisions1. This Regulation prescribes the procedures for the distribution and quality control of medicinal products (except for veterinary medicinal products), including homeopathic medicinal products and herbal medicinal products, as well as the procedures for and amount of wholesale data necessary for analysis of availability of medicinal products. [8 October 2013] 2. This Regulation shall apply to the medicinal products which: 2.1. have been manufactured industrially or prepared using a method which includes an industrial process; 2.2. in accordance with a prescription (formula magistralis) have been prepared at a pharmacy for an individual patient; 2.3. have been prepared at a pharmacy according to pharmacopoeial monographs and which are intended to be supplied directly to the patients who are served by the relevant pharmacy (formula officinalis); 2.4. a natural person imports, exports, sends by post or receives by post for personal use; 2.5. have been received or sent by postal consignments. It shall also apply to countries other than the European Union Member States and countries of the European Economic Area (hereinafter - the third countries); 2.6. have been given as a gift. [27 July 2010] 3. In free zones, free ports, special economic zones, customs warehouses, temporary storage places of goods, and also other places indicated or approved by the customs authority, the requirements for the distribution and quality control of medicinal products, and also the control and supervision measures laid down in this Regulation shall be applied. [17 March 2020] 4. [17 March 2020] 5. For the purpose of this Regulation: 5.1. suspicion of a defect exist when a notification which indicates that medicinal products do not have the quality specified in the registration documentation of the medicinal products has been received as regards the medicinal products; 5.2. medicinal products have a quality defect if the whole batch of medicinal products or a part thereof does not correspond to the prescribed requirements and the medicinal products are hazardous to the user, also if it is related to the packaging of the product; 5.3. a marketing authorisation holder is the person referred to in the laws and regulations regarding the procedures for registering medicinal products; 5.4. a recall of a batch are activities performed to withdraw a batch of medicinal products from the network of distribution and users; 5.5. the retail trade of medicinal products is the distribution of medicinal products to the public by a pharmacy; 5.6. the wholesale of medicinal products are all the activities which include the acquisition, storage, supply, or export of medicinal products (bringing out of medicinal products from the customs territory of a European Union Member State to the third countries), also if such activities take place in free zones, free ports, special economic areas, customs warehouses, or temporary storage places of goods and other places indicated or approved by the customs authority, except for the supply of medicinal products to inhabitants; 5.7. [8 October 2013]; 5.8. the giving of medicinal products as a gift is the distribution of medicinal products free of charge in compliance with the requirements specified in Paragraph 10 of this Regulation, regardless of which of the market participants is giving the medicinal products as a gift; 5.9. the brokering of medicinal products are all activities in relation to the sale or acquisition of medicinal products (except for the wholesale distribution of medicinal products) that do not include physical handling and that consist of negotiating independently and on behalf of another legal or natural person; 5.10. good distribution practice of medicinal products is a part of the quality assurance system which guarantees that the quality of medicinal products is preserved in all stages of the delivery chain from the manufacturing plant of medicinal products to a pharmacy or medical treatment institution, social care institution, practising veterinarians, practitioners, or institutions engaged in veterinary medical care; 5.11. repackaging is the movement of primary packaging of medicinal products from one secondary packaging to another secondary packaging, placement of the package leaflet in the secondary packaging or attaching thereof to the packaging of medicinal products (also by using a sticker), as well as attaching of a sticker or new labelling on the packaging or covering of the text part of the labelling; 5.12. actual non-availability of medicinal products is the situation when none of the manufacturers of medicinal products or medicinal product wholesale facilities actually have possibilities to supply medicinal products (there are no medicinal products in stock) to a pharmacy, medicinal treatment institution, social care institution, and also practising veterinarians and veterinary medical care institutions upon their request. In respect to the medicinal products that are included on the list of reimbursable medicinal products which is referred to in the laws and regulations regarding the procedures for the reimbursement of expenses for the acquisition of medicinal products and medical devices intended for out-patient medical treatment (hereinafter - the list of reimbursable medicinal products), supplies cannot be ensured within 24 hours, in respect to other medicinal products - within 48 hours; 5.13. artificial non-availability of medicinal products is the situation when a medicinal product wholesale facility refuses to supply particular medicinal products to a pharmacy although such medicinal products are available in the stocks of the relevant medicinal product wholesale facility, and also the situation when a pharmacy has not requested the particular medicinal products from the medicinal product wholesale facility in which they were available in accordance with the information published by the State Agency of Medicines. [27 July 2010; 8 October 2013; 3 December 2013; 2 February 2016; 17 March 2020] 6. The parallel import of medicinal products is the primary placement on the market of medicinal products, registered under the national registration procedure (also mutual recognition procedure and decentralised procedure) and delivered from a country of the European Economic Area for distribution in Latvia to a pharmacy, medical treatment institution, social care institutions, practising veterinarians, practitioners, and an institution engaged in veterinary medical care if they are placed on the market by such wholesaler of medicinal products which is not the manufacturer of such medicinal products, the marketing authorisation holder or their authorised representative (hereinafter - the parallel importer). [2 February 2016] 7. The parallel distribution of medicinal products is the placement on the market of medicinal products, registered within the centralised registration system and delivered from a country of the European Economic Area in Latvia if they are placed on the market by such wholesaler of medicinal products which is not the manufacturer of such medicinal products, the marketing authorisation holder or their authorised representative (hereinafter - the parallel distributor). [2 February 2016] 8. The centralised registration procedure is the registration of medicinal products pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency (hereinafter - Regulation No 726/2004 of the European Parliament and of the Council). 9. The requirements laid down in the Law on the Legal Trade of Narcotic and Psychotropic Substances and Medicinal Products, and also Precursors shall be complied with in respect of the deliveries of narcotic and psychotropic medicinal products from a country of the European Economic Area and to a country of the European Economic Area in addition to the requirements laid down in this Regulation. [4 March 2021] 10. Medicinal products given as a gift shall meet the following requirements: 10.1. medicinal products shall be given as a gift only to a medical treatment institution or a social care institution in accordance with the requirements of the laws and regulations regarding the procedures for the acquisition, storage, use and accounting of medicinal products in medical treatment institutions and social care institutions; 10.2. a written consent for the receipt of the particular medicinal products has been received from a medical treatment institution or a social care institution, specifying the name of medicinal products, strength or concentration, form, quantity in one packaging and the number of containers. If medicinal products are given as a gift to a medical treatment institution, the medicinal products given as a gift and the quantity thereof must conform to the profile of activities and volume of work of the medical treatment institution, and must be suitable for the treatment of the patients at the medical treatment institution. If medicinal products are given as a gift to a social care institution, medicinal products given as a gift must be suitable for the treatment of the patients at the social care institution; 10.3. only registered non-prescription medicinal products shall be given as a gift to social care institutions; 10.4. a medical treatment institution is permitted to receive the following as a gift: 10.4.1. such registered medicinal products that are not included on the list of reimbursable medicinal products (shall not apply to the medicinal products from List C of the list of reimbursable medicinal products when the medicinal products in the list of reimbursable medicinal products are intended for a patient with such diagnosis for which the acquisition of the medicinal products is not reimbursed); 10.4.2. unregistered medicinal products available for compassionate use in accordance with Article 83(2) of Regulation No 726/2004 of the European Parliament and of the Council (hereinafter - the medicinal products for compassionate use) within the scope of the programme referred to in Sub-paragraph 94.3 3.3 of this Regulation, only to such a medical treatment institution which is indicated in the relevant authorisation for the distribution of unregistered medicinal products. The donor of the medicinal products shall ensure that the conditions referred to in Article 83(8) of Regulation (EC) No 726/2004 of the European Parliament and of the Council are met; 10.5. the donor of medicinal products shall ensure that the patients who are treated with the given medicinal products are able to continue medical treatment with these medicinal products as long as it is necessary or until the moment when such medicinal products are included on the list of reimbursable medicinal products and are available within the scope of the reimbursement system for the acquisition of medicinal products; 10.6. at the moment of receipt of medicinal products, not less than one year shall remain until the expiry date of the medicinal products, but if the total period for use of the medicinal products is less than one year, not less than a half of the total period for use of the medicinal products shall remain until the expiry date of the medicinal products; 10.7. a bill of lading shall be drawn up for the medicinal products given as gifts; 10.8. the donor of the medicinal products shall ensure the collection and disposal of the unused medicinal products that were given as a gift in accordance with the laws and regulations governing the circulation of hazardous waste. [27 July 2010; 17 March 2020] II. Requirements for the Wholesale of Medicinal Products11. Such merchant or person registered with the State Revenue Service as a performer of economic activity (hereinafter - the performer of economic activity) shall be allowed to distribute medicinal products in wholesale to whom, in accordance with the laws and regulations regarding the procedures for the licensing of pharmaceutical activity, the State Agency of Medicines has issued: 11.1. a special authorisation (licence) for the opening (operation) of a medicinal product wholesale facility; 11.2. a special authorisation (licence) for the manufacturing or import of medicinal products which grants the authorisation holder the right to wholesale distribution of the medicinal products manufactured by it which the State Agency of Medicines has, when issuing the abovementioned special authorisation (licence), included in the database in conformity with the laws and regulations regarding the procedures for the licensing of pharmaceutical activity; 11.3. [27 July 2010]. [11 September 2012; 2 February 2016] 11.1 [8 October 2013] 11.2 [Paragraph shall come into force on 1 January 2023 and shall be included in the wording of the Regulation as of 1 January 2023. See Paragraph 171.19] 12. The person referred to in Paragraph 11 of this Regulation (hereinafter - the wholesaler of medicinal products) shall ensure compliance with the following requirements when distributing medicinal products in wholesale: 12.1. ensure that the premises, equipment, and installations are available to officials of the Health Inspectorate and the State Agency of Medicines for control at any time; 12.2. acquire medicinal products only from such persons (including registered in other countries) which have the right to distribute medicinal products in wholesale trade; 12.3. deliver medicinal products (or send them in a postal consignment) only to such persons which have the right to distribute medicinal products in wholesale trade and retail trade and to medical treatment institutions, social care institutions, practising veterinarians and veterinary medical care institutions which have the right to acquire the medicinal products in accordance with the laws and regulations regarding the procedures for the acquisition, storage, use, accounting, and destruction of medicinal products in medical treatment institutions and social care institutions, and the procedures by which a person engaged in veterinary medical practice shall perform activities with narcotic and psychotropic medicinal products, and the procedures by which a person engaged in veterinary medical practice shall acquire, store, and use medicinal products, and also to the persons for which the State Agency of Medicines has permitted to acquire medicinal products for ensuring their activity. Medicinal products shall be delivered without dividing the secondary packaging, except in the case when they have been repackaged; 12.4. develop an emergency situation plan in co-operation with the relevant manufacturer of the medicinal products or the owner of the medicinal products, or according to the order of the Health Inspectorate in order to ensure an effective recall of the medicinal products from the market; 12.5. record electronically each transaction involving medicinal products received and supplied, using the purchase and sale invoices, bills of lading, import or export declarations (if any), and also other documents, and ensure the possibility for the Health Inspectorate and the State Agency of Medicines to freely access electronically stored data without delay, and also to receive copies or printouts of such data. At least the following information shall be indicated on medicinal products, also medicinal products which are the subject-matter of the brokering of medicinal products, in the electronic recording system: 12.5.1. the name of the medicinal product; 12.5.2. the pharmaceutical form and strength or concentration; 12.5.3. the quantity in a packaging; 12.5.4. the date and time of the transaction when the medicinal product was received or sent; 12.5.5. the quantity of the medicinal product which is received, supplied or which is the subject-matter of the brokering of medicinal products; 12.5.6. the batch number of the medicinal product; 12.5.7. the term of validity of the medicinal product; 12.5.8. the manufacturer of the medicinal product; 12.5.9. the name and address of the consignee or supplier of the medicinal product, and also data for the identification thereof; 12.5.10. for medicinal products registered in Latvia and parallel imported medicinal products - the product number which the State Agency of Medicines assigns to each size of packaging of the medicinal product and which is indicated in the Medicinal Product Register of Latvia; 12.5.11. for medicinal products registered under the centralised registration procedure and parallel distributed medicinal products - the European Union number assigned by the European Medicines Agency to the size of the packaging of strength of each pharmaceutical form which is indicated as the product number in the Medicinal Product Register of Latvia; 12.5.12. for unregistered medicinal products - the identification number indicated in the authorisation referred to in Paragraphs 86 and 94 of this Regulation for the distribution of unregistered medicinal products for individually granted medicinal products; 12.5.13. the status of the medicinal product - is or is not on sale; 12.5.14. the medicinal products which have been sent back to a wholesaler by consignees of goods, including the name of the re-sender; 12.5.15. the price at which the medicinal product has been sold to the consignee of the medicinal products; 12.6. ensure that the data referred to in Sub-paragraph 12.5 of this Regulation are retained and available to officials of the Health Inspectorate and the State Agency of Medicines for at least five years, but in regard to narcotic and psychotropic medicinal products and substances - at least 10 years; 12.7. comply with the good distribution practice of medicinal products according to the guidelines for the good distribution practice of medicinal products published by the European Commission (available in the official language on the website of the State Agency of Medicines); 12.8. ensure proper and continuous delivery of medicinal products to pharmacies which have received a special authorisation (licence) for the opening (operation) of a pharmacy (hereinafter - the pharmacy) and to persons who are entitled to deliver medicinal products in order to ensure the needs of patients, as well as, by fulfilling obligations towards public service, to guarantee permanent access to a sufficient range of medicinal products and the delivery of any quantity of medicinal products ordered in conformity with the request in a short period of time, also in place of medicinal products which are withdrawn from the market, if the distribution of medicinal products has been suspended and the medicinal products are being withdrawn from the market in accordance with Sub-paragraph 115.1 of this Regulation. The wholesaler that specialises in sale and delivery of medicinal products of a particular manufacturer shall guarantee fulfilment of the duties specified in this Sub-paragraph in respect of the medicinal products of this manufacturer; 12.9. the wholesaler of medicinal products shall deliver as soon as possible medicinal products requested to be delivered urgently and present in the medicinal product stock by taking into account the distance to the place of delivery. Medicinal products that are included on the list of reimbursable medicinal products in accordance with the laws and regulations regarding the procedures for reimbursing expenses for the acquisition of medicinal products intended for medical treatment shall be delivered to a pharmacy upon a request of the pharmacy within 24 hours; 12.10. distribute only such medicinal products on the prices of which information has been provided in accordance with the laws and regulations regarding the principles for determining the price of medicinal products. This requirement shall not be applicable to unregistered medicinal products; 12.11. inform the Health Inspectorate and, if necessary, the marketing authorisation holder without delay if the medicinal products received or supplied are counterfeit or there are grounds for suspecting that they might be counterfeit; 12.12. create and maintain a quality system in which the duties, processes, and risk management measures are specified in relation to the type and volume of activity; 12.13. for all supplies of medicinal products a document shall be enclosed where the following is indicated: 12.13.1. the date of supply; 12.13.2. the supplied quantity (for each medicinal product); 12.13.3. the information referred to in Sub-paragraphs 12.5.1, 12.5.2, 12.5.10, 12.5.11, 12.5.12, and 12.5.15 of this Regulation; 12.13.4. the batch number of those non-prescription medicinal products which are included in Annex II to Commission Delegated Regulation (EU) 2016/161 of 2 October 2015 supplementing Directive 2001/83/EC of the European Parliament and of the Council by laying down detailed rules for the safety features appearing on the packaging of medicinal products for human use (hereinafter - Delegated Regulation No 2016/161) and prescription medicinal products which are included in Annex I to Delegated Regulation No 2016/161; 12.14. approve the official responsible for the conformity with good distribution practice who is a pharmacist with at least one year experience in wholesale trade of medicinal products or a person with the qualification which is specified for the responsible official in the laws and regulations regarding manufacturing control of medicinal products, and who has at least one year experience in the wholesale trade of medicinal products (these requirements shall be applicable also to a substitute of the responsible official); 12.15. indicate the information on the labelling and package leaflet for medicinal products which are distributed to a pharmacy, medical treatment institution, social care institutions, practising veterinarians, practitioners, and institutions engaged in veterinary medical care in the official language in accordance with the laws and regulations regarding the procedures for labelling medicinal products and the requirements to be brought forward for a package leaflet. For medicinal products which are distributed in wholesale trade in order to export them to a third country or to supply them to another country of the European Economic Area, it shall be clearly indicated in the accompanying document that "Medicinal products are intended for export to the third countries or for supply to another country of the European Economic Area". For parallel imported medicinal products, "Parallel imported medicinal products" shall be additionally indicated in the accompanying document; 12.16. make sure whether the received medicinal products are counterfeited by verifying the safety features on the outer packaging of the medicinal products which are referred to in Article 3(2)(a) and (b) of Delegated Regulation No 2016/161, and perform the obligations laid down for a wholesaler of medicinal products in accordance with Articles 10, 11, 12, 13, 19, 20, 21, 22, 24, Article 35(4), Articles 38, 40, and 42 of Delegated Regulation No 2016/161; 12.17. in conformity with Article 23 of Delegated Regulation No 2016/161, verify and delete the unique identifier located on the packaging of medicinal products from the Repository System of the Medicinal Products of Latvia when distributing: 12.17.1. vaccines for practices of family doctors, centres of paramedics, and centres of paramedics-midwives; 12.17.2. medicinal products for social care institutions; 12.17.3. medicinal products for persons who have an authorisation issued by the State Agency of Medicines in accordance with Section 48, Paragraph one of the Pharmaceutical Law; 12.17.4. medicinal products for practising veterinarians and veterinary medical care institutions; 12.17.5. medicinal products for the medical treatment institutions of prisons; 12.17.6. medicinal products for the State Emergency Medical Service, including when acquiring medicinal products for the State material reserves; 12.17.7. medicinal products for the medical treatment institutions of the National Armed Forces of the Ministry of Defence which acquire the medicinal products for the needs of civil defence and disaster management; 12.18. by using the information system of the State Agency of Medicines, each day until 10.00 o'clock, submit electronically information to the State Agency of Medicines in accordance with Sub-paragraphs 12.5.10, 12.5.11, and 12.5.12 of this Regulation, indicating the number of packages of the medicinal products in the stocks on the specific day and indicating separately the stocks intended for the market of Latvia and stocks which are intended for delivery to other European Union Member States or for export. [21 October 2008; 27 July 2010; 8 October 2013; 3 December 2013; 2 February 2016; 15 January 2019; 17 March 2020 / The new wording of Sub-paragraph 12.14 shall come into force on 1 July 2021. See Paragraph 171.16] 12.1 A merchant or a performer of economic activity who has been issued a special authorisation (licence) for the opening (operation) of a pharmacy shall distribute medicinal products to medical treatment institutions, social care institutions, practising veterinarians, and veterinary medical care institutions by ensuring compliance with the requirements laid down in Sub-paragraphs 12.1, 12.2, 12.3, 12.4, 12.5, 12.6, 12.13, 12.16, and 12.17 and Chapter III of this Regulation. [27 July 2010; 11 September 2012; 8 October 2013; 3 December 2013; 15 January 2019] 12.2 In order to fulfil the condition referred to in Sub-paragraph 12.2 of this Regulation, the wholesaler of medicinal products shall check, in cases where the medicinal products have been obtained from another wholesaler of medicinal products, whether the wholesaler of medicinal products who is a supplier complies with the good distribution practice specified in Sub-paragraph 12.7 of this Regulation and whether the supplier of medicinal products who is a medicinal product wholesale facility has the licence referred to in Sub-paragraph 11.1 or Paragraph 13 of this Regulation. If medicinal products are obtained from the manufacturer of medicinal products, the wholesaler of medicinal products shall check whether the manufacturer has the licence referred to in Sub-paragraph 11.2 of this Regulation for the manufacture of medicinal products or the licence referred to in Paragraph 13 of this Regulation. If medicinal products are obtained from the importer of medicinal products, the wholesaler of medicinal products shall check whether the importer has the licence referred to in Sub-paragraph 11.2 of this Regulation for the import of medicinal products. [8 October 2013; 3 December 2013] 12.3 [2 February 2016] 12.4 If the medicinal products are directly received from a third country and it is not intended to place them on the market in Latvia or another country of the European Economic Area, but the medicinal products are acquired in the third countries from persons who are entitled to supply medicinal products, the requirement referred to in Sub-paragraph 12.2 of this Regulation shall not be applied to the wholesaler of medicinal products. [8 October 2013; see Paragraph 171.6] 12.5 If any of the persons referred to in Sub-paragraph 12.17 of this Regulation verifies and deletes the unique identifier present on the packaging of medicinal products in the Repository System of the Medicinal Products of Latvia in accordance with Sub-paragraph 12.17 of this Regulation, then this requirement is not applied to the wholesaler of medicinal products. [15 January 2019] 12.6 The wholesaler of medicinal products shall ensure that the data referred to in Sub-paragraph 12.5 of this Regulation which are stored in electronic systems are comprehensible, legible, and easy to access, are protected against unauthorised access, data loss, or damage. The abovementioned data shall be duplicated or their backup copies shall be made, transferring the data to another storage system, and also auditing notes of such electronic data storage systems shall be kept. Data shall be stored for five years, and, upon request of the Health Inspectorate or State Agency of Medicines, they may be exported from the electronic system. [17 March 2020 / Paragraph shall come into force on 1 January 2021. See Paragraph 171.15] 12.7 The persons referred to in Paragraph 11.2 of this Regulation shall ensure the fulfilment of the requirements referred to in Sub-paragraph 12.1 of this Regulation and actions with medicinal products in accordance with the laws and regulations regarding the bringing in and out of medicinal products. [17 March 2020 / Paragraph shall come into force on 1 January 2021. See Paragraph 171.15] 13. The requirements laid down for the wholesaler of medicinal products in this Regulation shall apply to the wholesalers of medicinal products which distribute medicinal products in Latvia in accordance with the requirements of Section 25.1 of the Pharmaceutical Law (a special authorisation (licence) issued in another country of the European Economic Area for the wholesale trade of medicinal products or manufacture/import of medicinal products). 14. An undertaking registered in the country of the European Economic Area to which a special authorisation (licence) for the wholesale trade of medicinal products or manufacture/import of medicinal products has been issued in the home country shall, prior to the commencement of the medicinal product wholesale in the territory of the Republic of Latvia, provide the State Agency of Medicines with the following information: 14.1. the type, date of issue, number of the special authorisation (licence) granted in a country of the European Economic Area and the competent authority of the country which has granted the special authorisation (licence); 14.2. the firm name of the branch of a foreign merchant, the registration number within the Enterprise Register, the legal address and the address of the place of operation, telephone and fax number, electronic mail address of the undertaking; 14.3. the given name, surname, actual address of the place of employment, telephone and fax number and other means of communication of the official who is responsible for a good distribution practice. 15. The Health Inspectorate shall, in accordance with the conditions of Section 25.1 of the Pharmaceutical Law: 15.1. request information from the relevant foreign competent authority on the licence of the undertaking and ensure the exchange of information with foreign countries within the scope of its competence; 15.2. inform undertakings regarding the obligations towards public services. [21 October 2008] 16. The wholesaler of medicinal products shall appoint an official who is responsible for the fulfilment of the obligations towards public service and shall ensure that the abovementioned official is available (by telephone or using other means of communication) at any time of day or night, also, if in the case of emergency (spread of pathogens, toxins, chemical substances or nuclear radiation, during catastrophes, nature disasters, epidemics including pandemics), pharmacies or medical treatment institutions request medicinal products as a matter of urgency. 17. The medicinal product wholesale facility shall: 17.1. ensure an easy public access to the information on the distributable medicinal products and the prices thereof on its website; 17.2. notify the State Agency of Medicines of and indicate the website address of the medicinal product wholesale facility where information on medicinal products and their prices is available. [27 July 2010] 18. The persons referred to in Sub-paragraphs 11.1 and 11.2 and Paragraphs 12.1 and 13 of this Regulation shall notify the sales data of the medicinal products to the State Agency of Medicines (including on the parallel imported and parallel distributed medicinal products and medicinal products which are not intended to be placed on the market in Latvia but which are brought out to the European Union Member States or exported to third countries) in respect of each month until the fifteenth date of the following month or in respect of another period requested by the State Agency of Medicines. The following shall be indicated in the notification: 18.1. the name of the medicinal product, the form, strength or concentration of the medicinal product, the number in the package, the quantity of distributed packages, and the sales price of the medicinal product; 18.2. for medicinal products registered in Latvia and parallel imported medicinal products - the information referred to in Sub-paragraph 12.5.10 of this Regulation, for medicinal products registered under the centralised registration procedure and parallel distributed medicinal products - the information referred to in Sub-paragraph 12.5.11 of this Regulation; 18.3. for unregistered medicinal products - the information referred to in Sub-paragraph 12.5.12 of this Regulation; 18.4. the consignee of the medicinal products by including an indication "pharmacy", "medicinal product wholesale facility", "medical treatment institution", "veterinary medical care institution", "practising veterinarian", "practitioner", "exported", "brought out to a country of the European Economic Area" and "another consignee" (specifying if any). [27 July 2010; 11 September 2012; 8 October 2013; 3 December 2013; 2 February 2016; 17 March 2020] 19. [17 March 2020] 19.1 [17 March 2020] 20. Upon a request of the State Agency of Medicines, especially in relation to the pharmacovigilance system, the marketing authorisation holder shall submit to the State Agency of Medicines all data on the sales volume of the medicinal products and any other data owned (governed) thereby which apply to the quantity of the used medicinal products. [8 October 2013] II.1 Special Requirements for the Wholesale Trade in Medicinal Products for the Supply of Medicinal Products to Other European Union Member States or Export of Medicinal Products[17 March 2020 / Chapter shall come into force on 1 July 2020. See Paragraph 171.14] 20.1 The National Health Service shall publish on its website the medicinal products included on the list of reimbursable medicinal products in respect of which the National Health Service and marketing authorisation holder or wholesaler of medicinal products has entered into a contract for financial participation. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.2 The State Agency of Medicines shall supervise the availability of the medicinal products referred to in Paragraph 20.1 of this Regulation. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.3 It is prohibited to deliver or export the medicinal products referred to in Paragraph 20.1 of this Regulation to a European Union Member State if: 20.3 1. the marketing authorisation holder or the wholesaler of medicinal products has notified of the discontinuation of the delivery of the medicinal products which is applied until the time of renewal of the delivery of medicinal products; 20.3 2. the actual non-availability of the medicinal products has been established at a wholesale facility within the last three months. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.4 The State Agency of Medicines shall publish the information on the medicinal products which satisfy the conditions referred to in Paragraph 20.3 of this Regulation immediately, but not later than within one working day, on its website, indicating the name of the relevant medicinal product, its registration number, pharmaceutical form, strength, or concentration, and also in the case referred to in Sub-paragraph 20.3 1 of this Regulation - the foreseeable duration of the restriction which is specified according to the information provided by the marketing authorisation holder or wholesaler of medicinal products. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.5 The State Agency of Medicines may allow to supply the medicinal products referred to in Paragraph 20.3 of this Regulation to a European Union Member State or to export them: 20.5 1. if total stocks of medicinal products at wholesale facilities after the registered delivery or exportation will be available for at least one month (the amount of monthly consumption shall be calculated by taking into account the average consumption of the last three months); 20.5 2. in other cases due to special considerations by not causing availability risk for inhabitants. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.6 In order for the State Agency of Medicines to allow the delivery of the medicinal products referred to in Paragraph 20.4 of this Regulation to another European Union Member State or exportation thereof, the wholesaler of medicinal products shall submit a submission to the State Agency of Medicines. The following shall be indicated in the application: 20.6 1. the registration number of the medicinal product, the name of the medicinal product, the pharmaceutical form, strength, or concentration, the quantity in the package, the product number (according to the register of the State Agency of Medicines), the number of packages to be delivered or exported to another European Union Member State, and the number of packages in the stock at the time of notification; 20.6 2. the date when the delivery referred to in this Paragraph to another European Union Member State or exportation is intended; 20.6 3. the justification for ensuring the availability of the medicinal product to inhabitants. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.7 The State Agency of Medicines shall issue an authorisation to supply the medicinal products to another European Union Member State or to export them not later than within five working days from the moment of receipt of the submission referred to in Paragraph 20.6 of this Regulation. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] 20.8 The prohibition of supplies or export specified in this Chapter shall apply to the medicinal products which are intended for the market of Latvia. [17 March 2020 / Paragraph shall come into force on 1 July 2020. See Paragraph 171.14] III. Requirements for the Good Distribution Practice of Medicinal Products for a General or Open Type Pharmacy[17 March 2020] 21. The properties of medicinal products may not change during the distribution process of medicinal products. 22. A person to whom a special authorisation (licence) for the opening (operation) of a pharmacy has been issued shall establish a quality system in order to ensure that: 22.1. the medicinal products, which are distributed, are permitted in conformity with the legal acts of the European Community and the Republic of Latvia (those registered in the Medicinal Product Register of Latvia, registered within a centralised procedure or other country of European Economic Area, as well as permitted unregistered medicinal products); 22.2. the storage conditions of medicinal products are being observed at all times (also during the transportation of medicinal products) and they meet the requirements specified by the manufacturer; 22.3. the contamination of the medicinal products from the addition of or mixing with other products may not be possible; 22.4. the turnover of the stored medicinal products conforms to the requirements laid down in this Chapter; 22.5. the medicinal products are stored in safe and secure areas; 22.6. the ordered medicinal products are delivered to the addressee in accordance with Sub-paragraph 12.8 of this Regulation; 22.7. the procedures for determining any quality defects of the medicinal products are specified, and an effective recall procedure is provided. [8 October 2013; 3 December 2013] 23. [3 December 2013] 24. [3 December 2013] 25. Orders of medicinal products shall be addressed only to persons who hold the special authorisation (licence) referred to in Sub-paragraphs 11.1 and 11.2 of this Regulation or to the persons referred to in Paragraph 13 of this Regulation. 26. In addition to the conditions specified in the laws and regulations regarding operation of pharmacies, the manager of the pharmacy shall ensure the development of written procedures for the removal of medicinal products from the stock of medicinal products to be sold, returning and recall of medicinal products in accordance with the conditions referred to in Paragraph 31 of this Regulation, including in case of falsifications. [3 December 2013] 27. Descriptions of the procedures shall be approved by the manager of the pharmacy with a signature. [3 December 2013] 28. The manager of the pharmacy shall ensure that each acquisition and sale operation of medicinal products is recorded in minutes (records are made). The records (minutes) shall contain data by which all significant activities or events are traceable. The records (minutes) shall be retained for five years, and they shall be readily available. The information referred to in Sub-paragraph 12.5 of this Regulation shall be specified in the records (minutes). [3 December 2013] 29. The manager of the pharmacy shall ensure conformity with the following requirements: 29.1. premises, equipment and installations ensure proper conservation and distribution of medicinal products. Monitoring devices are calibrated and verified; 29.2. compliance with the following requirements is ensured in the receipt of medicinal products: 29.2.1. the receiving bay is arranged so that medicinal products are protected from weather influences during unloading. The reception area is separated from the storage area. It is checked whether the ordered medicinal products have been received and if the packaging of medicinal products is not damaged; 29.2.2. medicinal products subject to specific storage measures (for example, narcotic medicinal products, medicinal products requiring a specific storage temperature - immunological products) are immediately identified and stored in accordance with the description of procedures and requirements referred to in Sub-paragraph 29.3 of this Regulation; 29.3. implementation of the following requirements is ensured in storage of medicinal products: 29.3.1. medicinal products are stored in accordance with the temperature regime specified on the labelling and package leaflet of the medicinal products, taking into account that: 29.3.1.1. room temperature is from +15 ° to +25 ° Celsius; 29.3.1.2. a cool place is from +8 ° to +15 ° Celsius; 29.3.1.3. a cold place is from +2 ° to +8 ° Celsius; 29.3.2. thermolabile medicinal products (medicinal products which are sensitive to higher or lower temperatures) are stored in a cold room or a refrigerator, ensuring the appropriate temperature regime and its recording; 29.3.3. medicinal products that are sensitive to light are stored in places impervious to light (for example, tightly sealed containers, special packaging). If the medicinal products do not have special packaging impervious to light, they are stored in a dark place; 29.3.4. volatile substances and substances sensitive to moisture are stored in a cool place tightly sealed. Hygroscopic substances are stored in a dry room in hermetically sealed containers or plastic packaging, the container is sealed and paraffined, if necessary; 29.3.5. colourants and flavouring substances are stored apart from other medicinal products and substances (for example, within separate cupboards in tightly sealed packaging); 29.3.6. medicinal plants and herbs thereof are stored in a dry, well-ventilated room. Glass, paper or plastic packaging is used for the storage of medicinal plants and herbs thereof. Plants containing essential oils are stored in tightly sealed packaging; 29.3.7. the temperature regime in the medicinal products storage room is monitored and recorded periodically; 29.3.8. if medicinal products require a specific storage temperature (for example, immunological preparations), the storage area is equipped with temperature recorders or other devices that will indicate the periods when the specific temperature range has not been maintained. The control is adequate to maintain all parts of the storage area within such temperature range; 29.3.9. it is ensured that storage facilities are clean and free from litter, dust and pest, and relevant precautions are taken against spillage or packaging damage, micro-biological attack and cross contamination or mixing of medicinal products; 29.3.10. it is ensured that a system of stock rotation functions according to the principle that the product for which the term of validity expires first is distributed first (FEFO principle), as well as regular, periodic checks of system operation are carried out. Products beyond their expiry date or shelf life are stored apart from the usable stock, it is prohibited to either deliver or sell them; 29.3.11. if possible contamination is suspected, as well as medicinal products with a damaged seal or damaged packaging are withdrawn from the stock to be sold, and if not immediately destroyed, they are kept in a separated area so that they cannot be accidentally mixed in with other goods or sold in error. If necessary, such medicinal products are destroyed in accordance with the laws and regulations regarding hazardous waste disposal. [3 December 2013] 30. The manager of the pharmacy shall ensure that deliveries of medicinal products conform to the following requirements: 30.1. [3 December 2013]; 30.2. [3 December 2013]; 30.3. [3 December 2013]; 30.4. the medicinal products are transported in such a way that: 30.4.1. the identification thereof is not lost; 30.4.2. they do not contaminate or are not mixed together with other products or materials; 30.4.3. precautions are taken against their spillage, packaging damage or theft; 30.4.4. they are in a safe place (secure) and not subject to unacceptable temperature, light, moisture and other adverse influence, nor to microbiological attack and pests; 30.5. the medicinal products requiring controlled temperature storage are transported in accordance with the requirements laid down in Sub-paragraph 29.3.8 of this Regulation; 30.6. a person transporting medicinal products (a commercial carrier) takes the necessary precautionary measures to ensure that the vehicle is not used for illegal transport of medicinal products and informs the law enforcement authorities if suspicions have arisen that the vehicle is used illegally. [3 December 2013] 31. The manager of the pharmacy shall ensure conformity with the following requirements: 31.1. in case of returning medicinal products, if medicinal products do not have quality defects, they are stored in such a manner as to prevent re-distribution of such medicinal products until a decision has been reached on their use; 31.2. in order to recall medicinal products, the following activities are performed: 31.2.1. an emergency plan for the urgent recall of medicinal products and procedures for the recall of medicinal products is developed in writing, if the emergency recall is not urgent, as well as the person responsible for the execution and co-ordination of recalls is appointed; 31.2.2. any recall operations are recorded at the time they are carried out. The minutes are accessible to officials of the Health Inspectorate in the pharmacy; 31.2.3. the system for the recording of deliveries is developed so that all consignees of medicinal products could be immediately identified and contacted; 31.2.4. if medicinal products are recalled, the persons to whom they have been supplied are notified of the recall of medicinal products; 31.2.5. if medicinal products or a specific batch of medicinal products is recalled, according to the relevant class of quality faults the persons to whom they have been supplied are informed; 31.2.6. the medicinal products are recalled according to the notification approved by the marketing authorisation holder, the manufacturer of medicinal products or the Health Inspectorate; 31.2.7. the recalled medicinal products are immediately removed from the stock of medicinal products to be sold and are stored in a secure area, labelled with a notification "nav paredzēts pārdošanai" ["not for sale"]; 31.3. counterfeit medicinal products found in the distribution network are kept apart from other medicinal products to avoid any possibility of confusion. They are clearly labelled with a specific notification "nav paredzēts pārdošanai" ["not for sale"] and the marketing authorisation holder and the Health Inspectorate are immediately informed of them on the day of establishing the fact; 31.4. the return, rejection or recall of such medicinal products which are marked with a notification "nav paredzēts pārdošanai" ["not for sale"] and also receipt of counterfeit medicinal products is recorded at the time when the relevant operation is carried out. In each case, the decision to dispose these products shall be taken, and it shall be documented and recorded. [3 December 2013; 2 February 2016] 32. In order to monitor the fulfilment of and compliance with the good distribution principles, the manager of the pharmacy shall carry out self-control and shall record measures of the self-control. [3 December 2013] 33. [3 December 2013] IV. Requirements for the Parallel Import of Medicinal Products34. The parallel import of medicinal products is allowed only in cases when the parallel importer has obtained an authorisation for the distribution of parallel imported medicinal products within the Republic of Latvia in accordance with the requirements laid down in this Chapter and the abovementioned authorisation is valid. 34.1 A parallel importer shall request the information referred to in Parts II and II A of Annex 1 to this Regulation on parallel imported medicinal products from persons from whom medicinal products have been acquired in another country of the European Economic Area. The parallel importer which supplies the parallel imported medicinal products to another wholesaler of medicinal products, shall provide the information referred to in Parts II and II A of Annex 1 to this Regulation to such wholesaler of medicinal products. [2 February 2016] 35. A parallel importer shall notify, in accordance with Paragraph 39 of this Regulation, of its intention to distribute parallel imported medicinal products in Latvia to: 35.1. the marketing authorisation holder; 35.2. the State Agency of Medicines; 35.3. the owner of the trademark (brand) of medicinal products. [27 July 2010] 36. If the parallel imported medicinal products are being re-packaged, a parallel importer shall, upon a request of the owner of the trademark (brand) of medicinal products, deliver to him or her a sample of the re-packaged product. 37. The owner of the trademark (brand) of medicinal products may not use the right to the trademark (brand) of medicinal products to prohibit re-packaging if: 37.1. the right to the trademark (brand) used by the owner of the trademark (brand) in relation to the trading system developed by him or her promote artificial partitioning of the market between the European Union Member States; 37.2. the re-packaging does not negatively affect the original condition of the product; 37.3. the re-packager and manufacturer of the product are indicated on the new packaging; 37.4. the packaging intended for the distribution of the re-packaged product does not cause detriment to the trademark (brand) and does not harm the reputation of the owner thereof; 37.5. the owner of the trademark (brand) receives an advance notice prior to placing the product on the market. 38. Parallel imported medicinal products shall be registered (released for free circulation) in a country of the European Economic Area. Parallel import of medicinal products is allowed also, if a marketing authorisation holder recalls registration of the medicinal product registered in Latvia due to economic reasons which are not related to the safety, efficiency and quality of the medicinal products, if the authorisation referred to in Paragraph 34 of this Regulation for the distribution of parallel imported medicinal products in Latvia has been issued to the parallel importer and such authorisation is valid. [2 February 2016] 39. In order to obtain the authorisation referred to in Paragraph 34 of this Regulation, a parallel importer shall submit to the State Agency of Medicines a request submission for an authorisation for distribution of parallel imported medicinal products (hereinafter - the submission) in accordance with Annex 1 to this Regulation, containing a certification that the parallel importer has notified the relevant marketing authorisation holder and the owner of the trademark (brand) of medicinal products of the intention to commence the distribution of the parallel imported medicinal products. Information on the different types and sizes of the packaging of parallel imported medicinal products (with the same pharmaceutical form and the same strength of medicinal products), as well as different countries from which the parallel imported medicinal products are supplied, may be indicated in one submission. [2 February 2016] 40. The State Agency of Medicines shall, as soon as possible but not later than within five working days from the day of receipt of the submission at the State Agency of Medicines, perform primary expert-examination of the submitted information referred to in Paragraph 39 of this Regulation. It shall be checked in the primary expert-examination whether the submission has been drawn up in accordance with this Regulation and whether all documents have been submitted. The submitter of the submission shall be informed, without delay in electronic form, of the incomplete, incorrect information or information to be submitted in addition, as well as request for the competent authority of the country of origin of parallel imported medicinal products indicated in Annex 1 to this Regulation to submit the following information: 40.1. whether the particular medicinal product is registered (released for free circulation) in the country which is indicated in the request submission for an authorisation for the distribution of parallel imported medicinal products (Annex 1), and whether the registration of the medicinal product (marketing authorisation) is valid, as well as the (registration) number and date of issue of the abovementioned authorisation; 40.2. the name, legal address and address of the place of operation of the marketing authorisation holder; 40.3. the firm name, legal address and address of the place of operation of the medicinal product manufacturer, as well as information on the fact whether the licence of the manufacturer is valid; 40.4. the qualitative and quantitative composition of the medicinal product; 40.5. the shelf life of the medicinal product and the recommended storage conditions; 40.6. [2 February 2016]. [2 February 2016] 41. After completion of the primary expert-examination, the State Agency of Medicines shall check the submitted data and documents by taking into account the information received from another country of the European Economic Area (if any), shall compare them to the relevant data of medicinal products registered in Latvia, and shall assess them. Parallel imported medicinal products must meet the following requirements: 41.1. the parallel imported medicinal products are registered (a marketing authorisation is issued) in a country of the European Economic Area which is indicated in the request submission for an authorisation for the distribution of parallel imported medicinal products (Annex 1) as the country of origin; 41.2. the manufacturer of the parallel imported medicinal products (re-packager) has a special authorisation (licence) for the manufacture of medicinal products, and the manufacturing conforms to the requirements of good manufacturing practice; 41.3. the package leaflet and labelling (in the official language) for parallel imported medicinal products meet the regulations regarding procedures for labelling medicinal products and the requirements to be brought forward for package leaflets of medicinal products, and the leaflet is not misleading and provides accurate and complete information on the nature, composition, therapeutic effect, use and storage of the product; 41.4. the parallel imported medicinal products are identical to the medicinal products registered in Latvia or they possess only such permissible differences which are indicated in the laws and regulations regarding the procedures for labelling medicinal products and the requirements to be brought forward for package leaflets of medicinal products. The differences may not influence the therapeutic effect of a parallel imported medicinal product, pose a threat to public health or mislead patients: 41.4.1. the parallel imported medicinal products and the medicinal products registered in Latvia are manufactured using the same manufacturing methods, they have the same active substances and therapeutic effect. The manufacturer of parallel imported medicinal products and the manufacturer of medicinal products registered in Latvia, indicated in the submission referred to in Paragraph 39 of this Regulation, may be the same undertaking or undertakings within the scope of the same group of undertakings, or in case if they are independent undertakings, the manufacturer of parallel imported medicinal products and the manufacturer of medicinal products registered in Latvia have a contract with the same marketing authorisation holder; 41.4.2. the method of application introduced by the manufacturer of the medicinal products and the dose conform to the information on the use and dose within the registration documentation of medicinal products registered in Latvia; 41.4.3. the difference (if such exists) in the colouring agent (other colour code) is small; 41.4.4. the difference(-s), if any, is(are) clearly indicated in the labelling; 41.5. [2 February 2016]; 41.6. [2 February 2016]; 41.7. a parallel imported medicinal product is re-packaged in accordance with the laws and regulations regarding the manufacture of medicinal products, and the labelling thereof is drawn up in conformity with the laws and regulations regarding the procedures for labelling medicinal products and the requirements to be brought forward for package leaflets of medicinal products. [2 February 2016] 42. The State Agency of Medicines shall assess the conformity of parallel imported medicinal products with the requirements laid down in this Chapter and shall prepare an assessment report as soon as possible, as well as after the assessment of parallel imported medicinal products shall take the decision to issue an authorisation for the distribution of parallel imported medicinal products (Annex 2) or to refuse to issue an authorisation. An assessment report shall be prepared and the decision to issue an authorisation for the distribution of parallel imported medicinal products or to refuse to issue an authorisation shall be taken within the following time limits: 42.1. within one month from the day of receipt of the submission referred to in Paragraph 39 of this Regulation at the State Agency of Medicines; 42.2. within 15 days from the receipt of the submission referred to in Paragraph 39 of this Regulation at the State Agency of Medicines, if the parallel imported medicinal products are identical to medicinal products registered in Latvia (only difference in the size of the packaging of medicinal products is admissible) - they have the same name and strength, as well as the marketing authorisation holder and the manufacturer of medicinal products meet the requirements of Sub-paragraph 41.4.1 of this Regulation - or if the information on the packaging and the package leaflet is in the official language; 42.3. within five working days from the receipt of the submission referred to in Paragraph 39 of this Regulation at the State Agency of Medicines, if the relevant medicinal products registered in Latvia are not available on the market and if, in case of not receiving the medicinal products in due time, the life of a patient is endangered or significant harm to the health of the patient is caused. [2 February 2016; 17 March 2020] 43. If parallel imported medicinal products are different from the medicinal products registered in Latvia with excipients, the State Agency of Medicines may, if necessary, perform a solubility test thereof and request a description of the manufacturing method from the competent authority of the relevant country of the European Economic Area, informing the submitter of the relevant submission thereof in writing. The samples for testing shall be submitted, upon request of the State Agency of Medicines, by the recipient of the authorisation for the distribution parallel imported medicinal products. The expenses related to the sample testing of the medicinal product shall be covered by the parallel importer according to the price list of paid services of the State Agency of Medicines. [2 February 2016] 44. [17 March 2020] 45. The State Agency of Medicines shall take the decision to refuse to issue an authorisation for the distribution of parallel imported medicinal products if: 45.1. the submitted information does not meet the requirements laid down in this Chapter; 45.2. due to safety, efficacy and quality of the medicinal products the distribution of the medicinal products registered in Latvia is prohibited or distribution has been suspended, or the medicinal product has been removed from the market in Latvia or another country of the European Economic Area; 45.3. registration of the medicinal products registered in Latvia has been cancelled or suspended in accordance with the Pharmaceutical Law and the laws and regulations regarding the procedures for registering medicinal products. The abovementioned norm shall not apply to the case when the requester of registration has submitted the submission for the suspension of the registration of medicinal products and the reason is not related to risks to the health of inhabitants; 45.4. the marketing authorisation holder has revoked registration of the medicinal products registered in Latvia and the reason for revocation is related to risks to the health of inhabitants (safety, quality and efficacy of medicinal products); 45.5. the medicinal products have not been re-registered; 45.6. the re-registration of medicinal products has been refused. [11 September 2012] 46. If variations to the registration documentation for the medicinal products registered in Latvia have been approved in accordance with the laws and regulations regarding the procedures for registering medicinal products in respect of which an authorisation for the distribution of parallel imported medicinal products has been issued or the submission for the receipt of an authorisation has been submitted, the State Agency of Medicines shall inform the parallel importer and the submitter in writing: 46.1. of the approval of variations and distribution of the remaining stock of medicinal products in accordance with Paragraph 78.1 or 79 of this Regulation; 46.2. how the variations affect the dispensation of medicinal products (variation of classification), whether variations concern significant parts of the registration documentation of medicinal products which conform to I B or II type variations in the registration documentation or which are related to expanding registration in accordance with Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products (hereinafter - Regulation No 1234/2008 of the European Commission), and issue the approved package leaflet of medicinal products to the parallel importer for the registered medicinal products. [2 February 2016; 14 December 2021] 47. [11 September 2012] 48. The parallel importer shall: 48.1. after receipt of the information referred to in Paragraph 46 of this Regulation from the State Agency of Medicines, if variations in the package leaflet and labelling of medicinal products must be made, submit the submission (Annex 1) to the State Agency of Medicines, notifying of the introduction of variations to the package leaflet and labelling of the parallel imported medicinal products; 48.2. act according to the information provided by the State Agency of Medicines on the conditions for the distribution of the remaining stock of medicinal products; 48.3. [17 March 2020]; 48.4. submit the submission to the State Agency of Medicines, notifying of variations to the administrative data, contact information or size of the packaging of the medicinal products to be distributed; 48.5. follow the variations which are related to the medicinal products registered in the country which is indicated in the request submission for an authorisation for the distribution of parallel imported medicinal products (Annex 1) as the country of origin, and inform the State Agency of Medicines of variations which concern the medicinal products and the conditions for the distribution of the remaining stock of medicinal products. If variations refer to the information referred to in Part II, II A, or III of the submission (Annex 1), the submission for variations in the documentation shall be submitted to the State Agency of Medicines. [2 February 2016; 17 March 2020] 49. If the legal requisites of the owner of the authorisation for the distribution of parallel imported medicinal products change, he or she shall notify the State Agency of Medicines thereof in writing. The State Agency of Medicines shall decide on the issuing of a new authorisation in accordance with the procedures laid down in the Administrative Procedure Law. 50. The State Agency of Medicines shall determine a specific classification group to which the parallel imported medicinal products, conforming to the medicinal products registered in Latvia, shall belong in accordance with the laws and regulations regarding the procedures for the classification of medicinal products. 51. If belonging of the medicinal products registered in Latvia to the classification group referred to in Paragraph 50 of this Regulation is changed, the parallel imported medicinal products shall be distributed in conformity to the new medicinal product classification group specified for the medicinal products registered in Latvia. 52. The parallel importer shall: 52.1. after any operation related to medicinal product delivery from another country of the European Economic Area, make an accurate entry in a registration journal or another document intended for that purpose which confirms the origin of the parallel imported medicinal products, the batch number and quantity of the medicinal products brought in; 52.2. provide information to the Health Inspectorate and the State Agency of Medicines on the parallel imported medicinal products upon request of such authorities; 52.3. keep the medicinal product packaging material if the packaging has not been opened when re-packaging (re-labelling) the medicinal product; 52.4. keep one sample of each re-packaging operation product (a medicinal product, packaging material, package leaflet), if the packaging has been opened (for example, to change the outer packaging or package leaflet) when re-packaging (re-labelling) the medicinal product; 52.5. fulfil the requirements laid down in Articles 33, 40, and 42 of Delegated Regulation No 2016/161. The obligation laid down in Article 33 of Delegated Regulation No 2016/161 - to upload the information in the Repository System of the Medicinal Products of Latvia - shall be fulfilled by using the European repository system of medicinal products; 52.6. provide a notification to the State Agency of Medicines on the actual date of the commencement of the distribution (trade) of medicinal products in Latvia, indicating the product number referred to in Sub-paragraph 12.5.10 of this Regulation for each size of packaging of parallel imported pharmaceutical form, and shall, without delay, inform of the medicinal products which are permanently or temporarily not placed on the market of Latvia. [21 October 2008; 15 January 2019; 17 March 2020] 53. The State Agency of Medicines shall issue the authorisation in the form of an electronic document, sending it to the submitter of the submission to his or her electronic mail address within three working days if the relevant parallel importer has paid the specified fee for document expert-examination and performance of inspection according to the price list of paid services of the State Agency of Medicines. If the abovementioned person wants to receive the authorisation in the form of a printed document, it shall be issued within three working days for additional fee according to the price list of paid services provided by the State Agency of Medicines. [17 March 2020] 54. If the authorisation for the distribution of parallel imported medicinal products is refused or cancelled, the charge referred to in Paragraph 53 of this Regulation shall not be refunded. 55. The State Agency of Medicines shall take the decision to cancel the authorisation for the distribution of parallel imported medicinal products if: 55.1. the State Agency of Medicines cancels the relevant registration of the medicinal products registered in Latvia or the competent authority cancels the registration (marketing authorisation) in the country which is indicated as the country of origin in the request submission for an authorisation for the distribution of parallel imported medicinal products (Annex 1), for the parallel imported medicinal products due to such reasons as are associated with a threat to public health (safety, quality and efficacy of medicinal products) or prohibits supply of medicinal products and withdraws medicinal products from the market; 55.2. the State Agency of Medicines does not approve the variations of the parallel imported medicinal products; 55.3. manufacture of the parallel imported medicinal products does not conform to the requirements of the good manufacturing practice specified in the laws and regulations regarding the manufacture and control of medicinal products; 55.4. the authorisation holder has provided false information on the parallel imported medicinal products. [2 February 2016] 56. The State Agency of Medicines shall take the decision to suspend the authorisation for the distribution of parallel imported medicinal products if: 56.1. the State Agency of Medicines suspends the relevant registration of the medicinal products registered in Latvia or the competent authority suspends the registration (marketing authorisation) in the country which is indicated as the country of origin in the request submission for an authorisation for the distribution of parallel imported medicinal products (Annex 1), for the parallel imported medicinal products due to such reasons as are associated with a threat to public health (safety, quality and efficacy of medicinal products); 56.2. the authorisation holder has not submitted to the State Agency of Medicines data and documents on the variations or has not implemented the variations in accordance with the procedures laid down in this Regulation; 56.3. the manufacture of the parallel imported medicinal products does not conform to the requirements of the good manufacturing practice specified in the laws and regulations regarding the manufacture and control of medicinal products. [2 February 2016] 57. The State Agency of Medicines shall take the decision to cancel the suspension of the authorisation for the distribution of parallel imported medicinal products on the basis of the submission of the authorisation holder, if the reasons for suspending the operation of the authorisation have been eliminated. 58. The Health Inspectorate shall monitor and control the distribution of parallel imported medicinal products. [21 October 2008] 59. The State Agency of Medicines shall ensure that the competent authorities of the countries of the European Economic Area upon request receive information on the parallel imported medicinal products. V. Requirements for Parallel Distribution of Medicinal Products60. The parallel distributor shall, in accordance with Paragraph 60.1 of this Regulation, declare the intention to distribute the medicinal products registered in a centralised manner to: 60.1. the marketing authorisation holder and to the State Agency of Medicines, but if supplies are delivered to another Member State - to the competent authority of the relevant country; 60.2. the owner of the medicinal product trademark (brand). Prior to putting the re-packaged product on sale, as well as upon request of the owner of the medicinal product trademark (brand), he or she shall be supplied with a sample of the re-packaged product; 60.3. the European Medicines Agency in accordance with Paragraphs 63 and 64 of this Regulation. [27 July 2010; 8 October 2013] 60.1 The parallel distributor shall submit to the State Agency of Medicines a certification that: 60.1 1. it has notified the relevant marketing authorisation holder and the owner of the trademark of the medicinal products of the intention to commence the distribution of the specific parallel distributed medicinal products in Latvia, by indicating the particular addressee who has been notified and the date of providing the notification; 60.1 2. it has submitted to the European Medicines Agency the notification referred to in Paragraphs 63 and 64 of this Regulation for the specific parallel distributed medicinal products, and it shall indicate the date of submission thereof. [27 July 2010] 60.2 The parallel distributor shall submit to the State Agency of Medicines a notification on the actual date of commencing the distribution (sale) of medicinal products, indicating the identification number of the product referred to in Sub-paragraph 12.5.11 of this Regulation for each size of the packaging of parallel distributed pharmaceutical form, and shall, without delay, inform of the medicinal products which are permanently or temporarily not placed on the market of Latvia. [17 March 2020] 61. The owner of a medicinal product trademark (brand) shall not use trademark rights to prohibit re-packaging in the cases referred to in Paragraph 37 of this Regulation. 62. The distribution of parallel imported medicinal products is allowed also if the marketing authorisation holder registered in a centralised manner does not commence the distribution of such medicinal products in Latvia. 63. The parallel distributor shall submit a notification to the European Medicines Agency on the parallel distribution of centrally registered medicinal products (hereinafter - the notification) in conformity with the application form specified by the European Medicines Agency (published on the European Medicines Agency website) so that the European Medicines Agency may fulfil the requirements of Article 57(1)(o) of Regulation No 726/2004 of the European Parliament and of the Council. [27 July 2010; 8 October 2013] 63.1 A parallel distributor shall pay a fee to the European Medicines Agency for medicinal products which have been registered in accordance with Regulation No 726/2004 of the European Parliament and of the Council in order for the European Medicines Agency to check whether the conditions provided for in the marketing authorisation are conformed to. [8 October 2013] 63.2 A parallel distributor shall ensure the fulfilment of the requirements referred to in Sub-paragraph 52.5 of this Regulation. [15 January 2019 / Paragraph shall be applied from 9 February 2019 in conformity with the transitional measures specified Articles 48 and 50 of Delegated Regulation No 2016/161. See Paragraph 171.11] 64. If the annexes to the European Community marketing authorisation of medicinal products are amended or data in the information provided by the parallel distributor in the notification changes (for example, information on the re-packer or parallel distributor changes), the parallel distributor shall submit a notification on variations in the distribution of centrally registered parallel medicinal products in conformity with the application form specified by the European Medicines Agency (published on the European Medicines Agency website). This notification shall be submitted also in the case if the country in which it is intended to distribute the medicinal products is changed. [27 July 2010; 8 October 2013] 65. If a parallel distributor implements variations to the packaging of the medicinal products registered in a centralised manner, the variations shall be justified by the necessity to distribute the medicinal products. It is permitted to implement the following variations: 65.1. the information in the labelling and package leaflet is in the language of such country in which the medicinal products are offered for sale, and the abovementioned information is identical in all the languages used in the labelling and package leaflet; 65.2. the variations in the size of the packaging ensure that the offered packaging is included in the medicinal product marketing authorisation for the relevant medicinal products registered under the centralised registration procedure, issued by the European Community. 66. The Health Inspectorate shall monitor and control that the parallel distribution of medicinal products conforms to the requirements (labelling, package leaflet and batch identification) specified in the Annexes to the marketing authorisation of medicinal products from the European Community and that the notification provided by the European Medicines Agency which indicates the completion of notification examination is applied. [21 October 2008] V.1 Brokering of Medicinal Products[8 October 2013] 66.1 If medicinal products have been obtained via brokering, the wholesaler of medicinal products shall check whether the broker involved has fulfilled the requirements laid down in this Chapter. 66.2 The brokering of medicinal products may be carried out only by a person registered with the State Agency of Medicines. In order to register with the State Agency of Medicines, a person shall submit a submission to the State Agency of Medicines for registering a person who carries out transactions involving medicinal products (hereinafter - the registration submission) (Annex 2.1). At least the following information shall be indicated in the registration submission: 66.2 1. the given name and surname (performers of economic activity) or the firm name, legal address; 66.2 2. the address of the place of operation in Latvia; 66.2 3. the contact information in Latvia. 66.3 The person who brokers medicinal products shall notify the State Agency of Medicines of any variations in the data referred to in Paragraph 66.2 of this Regulation without delay, submitting the registration submission with the indicated variations (Annex 2.1). 66.4 The State Agency of Medicines shall register the person who brokers medicinal products by taking the decision to enter it in the register which is publicly accessible on the website of the State Agency of Medicines. The State Agency of Medicines shall indicate the data referred to in Sub-paragraphs 66.2 1 and 66.2 2 of this Regulation on the person who brokers medicinal products in the abovementioned register. 66.5 A registration certificate (Annex 2.2) issued by the State Agency of Medicines shall certify the fact of registration. The State Agency of Medicines shall send the registration certificate to the electronic mail address of the registered person within three working days after payment for the expert-examination of the documentation submitted for the issuance of a registration certificate and for the performance of the inspection. If the registered person wants to receive the registration certificate in the form of a printed document, it shall be issued within three working days for additional fee in accordance with the price list of paid services provided by the State Agency of Medicines. [2 February 2016; 17 March 2020] 66.6 The persons brokering medicinal products shall: 66.6 1. ensure that the medicinal products which are the subject-matter of brokering are registered in Latvia or under the centralised registration procedure referred to in Paragraph 8 of this Regulation; 66.6 2. create and maintain a quality system in which the duties, processes, and risk management measures are specified in relation to the type and volume of their activity. 66.7 The requirements laid down in Sub-paragraphs 12.4, 12.5, 12.6, 12.7 and 12.11 of this Regulation shall also be applied to the brokering of medicinal products. 66.8 If the person brokering medicinal products does not meet the requirements referred to in Paragraphs 66.3, 66.6 and 66.7 of this Regulation, the State Agency of Medicines is entitled to take the decision to exclude the relevant person from the register referred to in Paragraph 66.4 of this Regulation. The State Agency of Medicines shall notify the abovementioned person of the decision to exclude from the register in accordance with the procedures laid down in the Law on Notification. VI. Special Requirements for Distribution of Medicinal Products in Pharmacies67. A pharmacy (a retailer of medicinal products) shall, in the distribution of medicinal products, ensure fulfilment of the requirements referred to in Sub-paragraphs 12.1, 12.2, 12.4, 12.5, 12.6, 22.1, 22.2, 22.3, 22.5, 22.6, 22.7, 29.1, 29.2, 29.3, 31.3 and 31.4 of this Regulation. [27 July 2010] 67.1 Medicinal products shall not be placed in the part of the customer servicing area where customers are present. [4 August 2009 / Paragraph shall come into force on 1 September 2009. See Paragraph 171.1] 67.2 A general type pharmacy shall be regarded to be the person referred to in Delegated Regulation No 2016/161 which is allowed or which is entitled to deliver medicinal products to inhabitants and it shall fulfil the obligations laid down in Articles 10, 11, 13, 25, 27, 28, 29, and 30 of Delegated Regulation No 2016/161, including verify packagings of medicinal products also for unregistered medicinal products with safety features. A general type pharmacy may verify and delete the unique identifier present on the packaging of medicinal products from the Repository System of the Medicinal Products of Latvia, delivering medicinal products with safety features to its branch of pharmacy. [15 January 2019; 17 March 2020] 68. A pharmacy is allowed to acquire medicinal plants (herbs) from the public and to distribute them to the public, if the head of the pharmacy has examined the medicinal plants (herbs) that have been acquired and has determined the conformity thereof to the description of the permitted pharmacopoeia monograph, as well as, if the place of medicinal plant harvesting and conditions of herb preparation are known, to the head of the pharmacy. 69. Unregistered medicinal products may be dispensed in a pharmacy to inhabitants: 69.1. on the basis of a prescription completed in accordance with the laws and regulations regarding the writing out of prescriptions; 69.2. without a prescription if the medicinal products are included in the authorisation for distribution referred to in Paragraph 86 of this Regulation and the State Agency of Medicines has determined inclusion of the particular medicinal products in the category of medicinal products not subject to medical prescription in accordance with the laws and regulations regarding the procedures for the classification of medicinal products. [27 July 2010] 70. Medicinal products shall be dispensed in conformity with the quantity indicated in the prescription. If parallel imported medicinal products are different from the medicinal products registered in Latvia, when dispensing the abovementioned medicinal products in a pharmacy, the patient and doctor (by telephone) shall be informed of the difference between the parallel imported medicinal products and the medicinal products registered in Latvia. 70.1 When dispensing medicinal products for a prescription written out by a doctor, the pharmacists shall ascertain that the patient will be able to use the medicinal products dispensed, using them according to the dosage and frequency of use indicated by the physician, before expiry of their term of validity. When dispensing non-prescription medicinal products, the pharmacist shall inform the patient of the term of validity of medicinal products if less than two months have left until expiry of the term of validity. [2 February 2016] 71. If a pharmacy does not have medicinal products in stock, the pharmacy shall accept the request of a patient, and also medicinal treatment institution, social care institution, practising veterinarian, or veterinary medical care institution, including if the opinion of the relevant professional association of doctors or professional panel of veterinarians has been appended thereto, and on the same day after receipt of the request shall order the necessary medicinal products, using electronic mail or electronic systems for ordering medicinal products at least with one of the persons referred to in Paragraphs 11 and 13 of this Regulation which has such medicinal products in stock in accordance with the information available on the website of the State Agency of Medicines. It shall be regarded that the persons referred to in Paragraphs 11 and 13 of this Regulation have received such order of medicinal products on the same day. In respect of unregistered medicinal products, the pharmacy shall ensure the implementation of the requirements laid down in Paragraph 72 of this Regulation. After receipt of the medicinal products, the pharmacy shall: 71.1. immediately deliver the medicinal products or inform of the receipt of medicinal products the medical treatment institution, social care institution, practising veterinarian or institution engaged in veterinary medical care which has requested the medicinal products; 71.2. notify the patient who has ordered the medicinal products of the receipt thereof. [27 July 2010; 8 October 2013; 17 March 2020] 71.1 The persons referred to in Paragraphs 11 and 13 of this Regulation shall provide a reply to the pharmacy to the request referred to in Paragraph 71 of this Regulation, using electronic mail, not later than within 12 hours after receipt of the order for the medicinal products which are included on the list of reimbursable medicinal products and not later than within 24 hours for other medicinal products. A pharmacy shall notify the Health Inspectorate of the refusal to deliver medicinal products if it is apparent from the information available on the website of the State Agency of Medicines that such medicinal products are in the stock of the person referred to in Paragraphs 11 and 13 of this Regulation on the day of sending the request, indicating the name, strength, concentration, quantity of packagings of the requested medicinal products, and also the date of the request and the person referred to in Paragraphs 11 and 13 of this Regulation from which the refusal has been received, including the justification of the refusal (if any has been provided). [17 March 2020] 71.2 A pharmacy shall store the information referred to in Paragraph 71.1 of this Regulation which is comprehensible, legible, and easy to access for one year. The officials of the Health Inspectorate have the right, at any time without delay, to access such data without hindrance and also to make copies of such data. Such data shall be protected against unauthorised access, data loss, or damage. The abovementioned data shall be duplicated or backup copies thereof shall be created and transferred to another storage system. [17 March 2020] 71.3 If it is not possible to get the requested medicinal products, a pharmacy shall notify thereof a person or institution which has ordered the medicinal products. [17 March 2020] 72. The pharmacy shall comply with the following requirements in the distribution of unregistered medicinal products: 72.1. the pharmacy shall accept and register the prescription or the request of an institution and practising veterinarian referred to in Paragraph 71 of this Regulation if unregistered medicinal products are not in the stocks of the pharmacy; 72.2. contact the person referred to in Paragraph 11 or 13 of this Regulation as regards the delivery of unregistered medicinal products and submit to the abovementioned person a written request of the pharmacy for the delivery of medicinal products. The prescription referred to in Sub-paragraph 72.1 of this Regulation and the request of an institution and practising veterinarian need not be appended to the request of the pharmacy. The pharmacy shall indicate the following in the request: 72.2.1. the name of the unregistered medicinal products to be delivered, the form, strength or concentration of the medicinal products, the quantity in the packaging unit, the quantity of the packaging units, the time limit for the delivery of the medicinal products, the name, licence number, contact information of the pharmacy, and the request number of the pharmacy; 72.2.2. the person who has written out the prescription, and the medical treatment institution, social care institution, institution engaged in veterinary medical care and practising veterinarian who requests the medicinal products; 72.2.3. the manufacturer of the medicinal products, if necessary; 72.3. is permitted not to provide a written request to the wholesaler of medicinal products for the medicinal products which: 72.3.1. correspond to the list of medicinal products specified by the National Health Service in accordance with the laws and regulations regarding the procedures for the acquisition, storage, use, accounting and destruction of medicinal products in medical treatment institutions and social care institutions, and which are required for the provision of inpatient health care services paid from the State budget (hereinafter - the list of usable medicinal products) (shall apply to the request of an institution and practising veterinarian); 72.3.2. are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment in accordance with the laws and regulations regarding the procedures for the reimbursement of expenses for the acquisition of medicinal products and medical devices intended for out-patient medical treatment (hereinafter - the medicinal products which are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment) (shall apply to the request of the pharmacy); 72.4. store the prescription and also the request of the institution and practising veterinarian at the pharmacy. [27 July 2010; 11 September 2012; 2 February 2016] 72.1 A pharmacy shall, upon receipt of prescriptions written out in the European Union, countries of the European Economic Area and the Swiss Confederation, comply with the following requirements: 72.1 1. ascertain whether the prescription form contains at least the following specified elements: 72.1 1.1. information on the patient - the given name, surname (written in full names without using initials), the date of birth and the address; 72.1 1.2. information on the prescriber of the medicinal products - the given name, surname (written in full names without using initials), address, telephone, fax, e-mail, speciality, identification number and signature of the physician who wrote out the prescription; 72.1 1.3. the date of writing out the prescription; 72.1 1.4. indicated international nonproprietary name of the medicinal products or, if none, the generally accepted name of the medicinal products, or if the doctor has indicated the reasons for substitution prohibition or has prescribed medicinal products of biological origin - the name assigned by the manufacturer of medicinal products; 72.1 1.5. information on the pharmaceutical form (pills, liquid, ointment, etc.), quantity, strength, and dosage; 72.1 2. when dispensing medicinal products for a prescription written out in the European Union, countries of the European Economic Area and the Swiss Confederation, the pharmacist shall issue accurate information (a cashierʼs cheque or a receipt) in which the name, strength, quantity, price of the issued medicinal products and the date of issuance is indicated; 72.1 3. a prescription written out in the European Union, countries of the European Economic Area and the Swiss Confederation may be refused in a pharmacy: 72.1 3.1. if there are justified suspicions of its authenticity; 72.1 3.2. if the content of the prescription is incomprehensible, including, it is impossible to identify the medicinal products prescribed or the instructions for the use of medicinal products; 72.1 3.3. if there are other ethical reasons which preclude from dispensing the medicinal products to the patient and conform to the activities which are necessary and commensurate with human health protection and are not discriminatory; 72.1 4. recognition of a prescription written out in the European Union, countries of the European Economic Area and the Swiss Confederation shall not apply to prescriptions which prescribe narcotic or psychotropic medicinal products, and also narcotic analgesics; 72.1 5. after medicinal products are dispensed, the prescription remains at the pharmacy. If the patient requests, the pharmacy shall issue a copy of the prescription. [3 December 2013] 73. When medicinal products have been dispensed (except for an ordinary prescription on which the prescribed medicinal products may be dispensed repeatedly), as well as the requests of the institution and practising veterinarian shall remain at the pharmacy. [27 July 2010] 74. A pharmacy shall, in accordance with the laws and regulations regarding the distribution of narcotic and psychotropic medicinal products, register in the strict account register for narcotic and psychotropic medicinal products equivalent thereto such medicinal products and substances for medicinal products to be manufactured which contain the following substances: 74.1. atropine sulphate; 74.2. silver nitrate; 74.3. arsenous acid anhydride; 74.4. crystalline sodium arsenate; 74.5. tetracaine hydrochloride (dicaine); 74.6. narcotic analgesics which have been recognised as such by the State Agency of Medicines. [27 July 2010] 75. The request of an institution and practising veterinarian for the dispensing of medicinal products shall be stored at a pharmacy for three years. Bills of lading regarding the circulation of the substances and medicinal products referred to in Paragraph 74 of this Regulation shall be drawn up and kept and reports thereon shall be provided in accordance with the laws and regulations regarding the trade of narcotic and psychotropic medicinal products. [27 July 2010] 76. [6 March 2018] 77. The distribution of medicinal products in vending (sales) machines is prohibited. VII. Conditions for Distribution of Remaining Stock of Medicinal Products after Confirmation of Variations to Registration Documentation78. A marketing authorisation holder is entitled to introduce variations in registered medicinal products after the decision to approve variations to the registration documentation thereof has entered into effect in accordance with the laws and regulations regarding the procedures for the registration of medicinal products. The marketing authorisation holder shall be responsible for the compliance with the time limit for the introduction of variations indicated on the submission form. A marketing authorisation holder shall introduce the relevant variations in the registered medicinal products from the next manufacturing if: 78.1. the manufacture and distribution of medicinal products have not been commenced previously (medicinal products have not been previously placed on the market); 78.2. time limit for the distribution of the remaining stock of medicinal products has been determined concurrently with the approval of variations or the distribution of the remaining stock of medicinal products is not allowed (for example, urgent restrictions related to the safety of medicinal products, public health protection, or safe use of medicinal products). [14 December 2021] 78.1 Wholesalers of medicinal products and pharmacies are entitled to distribute, but medical treatment institutions and social care institutions, veterinary medical care institutions or practising veterinarians are entitled to use the medicinal products referred to in Paragraph 78 of this Regulation (hereinafter - the remaining stock of medicinal products) until expiry of the term of their validity, except when the State Agency of Medicines: 78.1 1. has determined a time limit for the distribution of the remaining stock of medicinal products; 78.1 2. does not allow the distribution of the remaining stock of medicinal products if variations are related to urgent restrictions related to the safety of medicinal products, public health protection, or safe use of medicinal products (for example, variations in therapeutic indications, contraindications, or warnings, new information on the safe use of medicinal products has been obtained). [14 December 2021] 79. The State Agency of Medicines shall, without delay, notify the decision referred to in Sub-paragraphs 78.1 1 and 78.1 2 of this Regulation to the marketing authorisation holder, parallel importer, and the Health Inspectorate, and also publish it on its website. [14 December 2021] 80. After the decision to approve variations to the registered medicinal products has entered into effect in accordance with the laws and regulations regarding the procedures for the registration of medicinal products, the marketing authorisation holder shall ensure the introduction of variations in the labelling and package leaflet in conformity with introduction of variations in accordance with Article 24 of European Commission Regulation (EC) No 1234/2008. [17 March 2020] 81. If the marketing authorisation holder does not re-register the medicinal products and asks to cancel the registration of medicinal products and to exclude the medicinal products from the Medicinal Product Register of Latvia due to reasons not related to safety, quality, and efficiency of medicinal products, the remaining stock of medicinal products for which batch release has been previously performed may be distributed for six months after entering into effect of the decision to cancel the registration of medicinal products and to exclude them from the Medicinal Product Register of Latvia or until expiry of the term of validity of medicinal products if the remaining term of validity is shorter than six months. If the re-registration of medicinal products is refused or the registration of medicinal products is cancelled or suspended due to reasons related to safety, quality, and efficiency of medicinal products, the State Agency of Medicines may, by assessing the public health risks, decide on the time limit for the sale of the remaining stock of medicinal products in accordance with the laws and regulations regarding the procedures for registering medicinal products. The State Agency of Medicines shall also inform the parallel importer of the decision taken. [17 March 2020] 81.1 If the authorisation for the distribution of parallel imported medicinal products is suspended or cancelled due to reasons not related to safety, quality, and efficiency of medicinal products, the remaining stock of medicinal products may be distributed for six months after entering into effect of the relevant decision of the State Agency of Medicines to suspend or cancel the authorisation for the distribution of parallel imported medicinal products or until expiry of the term of validity of medicinal products if the remaining term of validity is less than six months. If the authorisation of parallel imported medicinal products is suspended or cancelled due to reasons related to safety, quality, and efficiency of medicinal products, the State Agency of Medicines shall indicate the time limit for the sale of the remaining stock of medicinal products, assessing the information provided by the parallel importer on the remaining stock of medicinal products and public health risks. [17 March 2020] 82. Upon request of the State Agency of Medicines, marketing authorisation holders, manufacturers of medicinal products, medicinal product wholesale facilities, pharmacies, medical treatment institutions, practising veterinarians, veterinary medical care institutions, and social care institutions shall provide the State Agency of Medicines and the Health Inspectorate with a notification on the remaining stocks of medicinal products (that shall also apply to the medicinal products for parallel import and parallel distribution) in which the approved variations to registered medicinal products have not been introduced in accordance with the laws and regulations regarding the procedures for the registration of medicinal products in conformity with the request of the Agency, indicating the series number and quantity of the particular batch of medicinal products. [17 March 2020] VIII. Requirements for Distribution of Unregistered Medicinal Products83. The Health Inspectorate, but in respect of medicinal products for human use which are intended to be used by animals - the Food and Veterinary Service, is entitled to verify the justification for the writing of prescription for unregistered medicinal products. [21 October 2008; 27 July 2010] 84. If doubts or suspicion arise of the distribution of unregistered medicinal products, the State Agency of Medicines is entitled to request the Health Inspectorate to verify the justification for the writing of prescriptions for unregistered medicinal products, but in respect of medicinal products which are intended to be used by animals - the Food and Veterinary Service. [21 October 2008; 27 July 2010] 85. A doctor, medical treatment institution, as well as a practising veterinarian and an institution engaged in veterinary medical care shall, before prescribing and designating unregistered medicinal products to a patient (shall not apply to medicinal products for which the authorisation referred to in Paragraph 86 of this Regulation for the distribution of medicinal products registered in a country of the European Economic Area but not registered in the Republic of Latvia has been issued) shall obtain information on properties of such medicinal products to evaluate the therapeutic efficacy and safety thereof for a patient and to ensure monitoring of their use, as well as shall be responsible for prescribing or designating such medicinal products. [27 July 2010; 8 October 2013] 86. In order to receive an authorisation for the distribution of medicinal products registered in a country of the European Economic Area but not registered in the Republic of Latvia (Annex 4), a holder of a marketing authorisation issued in a country of the European Economic Area or a submitter of the registration submission (if the medicinal products are in the process of registration) shall submit the submission (Annex 5) to the State Agency of Medicines. [27 July 2010; 11 September 2012] 86.1 The State Agency of Medicines is entitled to take the decision to issue the authorisation referred to in Paragraph 86 of this Regulation, also upon proposal of the National Health Service or a medical treatment institution, and also to put forward a proposal itself to issue the authorisation on the basis of public health considerations if the submission referred to in Paragraph 86 of this Regulation has not been submitted to the State Agency of Medicines. [27 July 2010; 11 September 2012] 86.2 The State Agency of Medicines shall issue the authorisation referred to in Paragraph 86 of this Regulation in the name of the holder of the marketing authorisation issued in a country of the European Economic Area or, if medicinal products are in the process of registration in a country of the European Economic Area, the relevant marketing authorisation requester. [27 July 2010] 86.3 The State Agency of Medicines shall take the decision to issue the authorisation referred to in Paragraph 86 of this Regulation for the definite period - five years. The abovementioned decision shall become invalid if the medicinal products referred to in Paragraph 86 of this Regulation are being registered in Latvia and included in the Medicinal Product Register of Latvia. [27 July 2010; 2 February 2016] 86.4 Five years after taking the decision referred to in Paragraph 86.3 of this Regulation, the State Agency of Medicines is entitled to take the decision to issue the authorisation referred to in Paragraph 86 of this Regulation for an unlimited period, except when, on the basis of the evidence acquired in pharmacovigilance, the State Agency of Medicines has taken the decision to issue the relevant authorisation for a definite period - five years. The abovementioned decision shall become invalid if the medicinal products specified in the authorisation are being registered in Latvia and included in the Medicinal Product Register of Latvia. [27 July 2010; 8 October 2013; 2 February 2016] 87. Prior to the issuance of the authorisation for the distribution of unregistered medicinal product referred to in Paragraph 86 of this Regulation, the State Agency of Medicines shall, by taking into account public health considerations: 87.1. notify the marketing authorisation holder in the country of the European Economic Area of the proposal to grant an authorisation for the distribution of the relevant medicinal products in the Republic of Latvia; 87.2. request the competent authority of the relevant country of the European Economic Area to submit a copy of the medicinal product assessment report and a valid marketing authorisation for the relevant medicinal products; 87.3. inform the authorisation holder of the requirements for the advertising, labelling and package leaflet of medicinal products; 87.4. inform the authorisation holder of the requirements in the monitoring of the adverse effects caused by the medicinal product use. [27 July 2010] 87.1 The State Agency of Medicines shall take the decision to grant the authorisation after receipt of the medicinal product assessment report provided by the competent authority of the relevant country of the European Economic Area and the medicinal product marketing authorisation. If the competent authority of the country of the European Economic Area has not approved the medicinal product assessment report, the State Agency of Medicines shall request the relevant competent authority of the country of the European Economic Area to submit the package leaflet of the relevant registered medicinal products and a summary of product characteristics in the original language or in English if the package leaflet and summary of product characteristics have been approved in English. In such case, the State Agency of Medicines shall take the decision to grant an authorisation after receipt of the marketing authorisation of medicinal products, the package leaflet of medicinal products, and the summary of product characteristics. If the authorisation referred to in Paragraph 86 of this Regulation is requested by a marketing authorisation holder or the submitter of the registration submission (if the medicinal products are within the process of registration), the State Agency of Medicines shall request to submit a certified translation of the package leaflet of medicinal products and of the summary of product characteristics in accordance with the laws and regulations regarding the procedures for certifying translations of documents in the official language. [11 September 2012] 87.2 If a request of the competent authority of another country of the European Economic Area has been received at the State Agency of Medicines for the submission of a copy of the medicinal product assessment report on medicinal products registered in Latvia and a valid registration certificate of the relevant medicinal products, the State Agency of Medicines shall submit the copy of the assessment report and the marketing authorisation of the relevant medicinal products within 30 days after receipt of the request. [8 October 2013] 88. A holder of the authorisation referred to in Paragraph 86 of this Regulation or a submitter of the registration submission (if the medicinal products are within the process of registration) shall, in accordance with the laws and regulations regarding procedures for labelling medicinal products, submit a labelled packaging and a package leaflet in the official language to the State Agency of Medicines. [11 September 2012] 89. When issuing the authorisation referred to in Paragraph 86 of this Regulation, the State Agency of Medicines shall determine a classification group to which the medicinal products shall belong in accordance with the laws and regulations regarding the procedures for the classification of medicinal products. [11 September 2012] 89.1 When issuing the authorisation referred to in Paragraph 86 of this Regulation for the distribution of unregistered medicinal products, the State Agency of Medicines shall approve the labelling and package leaflet of the medicinal products in the official language by consulting, if necessary, with the marketing authorisation holder, manufacturer and other competent authorities. [27 July 2010] 89.2 The wholesaler of medicinal products which delivers the medicinal products shall ensure that the distributable medicinal products have labelling and package leaflet in the official language in accordance with the laws and regulations regarding the procedures for labelling medicinal products which stipulate the requirements for medicinal products and the package leaflet. [27 July 2010] 90. The holder (owner) of the authorisation referred to in Paragraph 86 of this Regulation shall ensure: 90.1. the supervision of side effects which are caused by the use of the medicinal products in accordance with the laws and regulations regarding the procedures for pharmacovigilance, analogous to that specified for registered medicinal products; 90.2. the compliance with the requirements specified in the laws and regulations regarding the advertising of medicinal products. [27 July 2010; 11 September 2012; 8 October 2013] 91. On the website address of the State Agency of Medicines, the State Agency of Medicines shall ensure publicly accessible information on unregistered medicinal products to which, in accordance with Paragraph 86 of this Regulation, an authorisation for the distribution of unregistered medicinal products has been issued in accordance with Sub-paragraph 149.7 of this Regulation. [27 July 2010; 8 October 2013] 92. The State Agency of Medicines shall notify the European Commission of medicinal products not registered in Latvia for which, in accordance with Paragraph 86 of this Regulation, an authorisation for the distribution of unregistered medicinal products has been issued and medicinal products for which the authorisation for the distribution of unregistered medicinal products is cancelled, and shall indicate the given name, surname or firm name and address of the authorisation holder. 93. The authorisation referred to in Paragraph 86 of this Regulation gives the wholesaler of medicinal products referred to in Paragraphs 11 and 13 of this Regulation the right to distribute the medicinal products in wholesale trade, while the merchant or performer of economic activity who has been issued a special authorisation (permit) for the opening (operation) of a pharmacy - to distribute the medicinal products in accordance with Paragraph 12.1 of this Regulation. [11 September 2012; 8 October 2013] 94. The wholesale distribution of medicinal products not registered in Latvia for which the authorisation referred to in Paragraph 86 of this Regulation has not been issued, if the following has been received: 94.1. the authorisation for the distribution of unregistered medicinal products for individually granted medicinal products (applies to medicinal products registered and used in other countries): 94.1.1. to a clearly known patient or individual patients (Annex 6), by fulfilling the bona fide request, and to the person who has prescribed or assigned the medicinal products, taking responsibility for prescribing or assigning the medicinal products; 94.1.2. on the basis of the decision of the Minister for Health referred to in Section 10, Clause 7, Sub-clause "c" of the Pharmaceutical Law, if the medicinal products are required for the provision of medical assistance in case of an emergency, natural disaster or epidemics, as well as for eliminating possible or already detected diffusion of pathogenic, toxic, chemical substances or radiation which could cause harm; 94.2. the authorisation for the distribution of unregistered medicinal products for individually granted medicinal product for compassionate use (Annex 6). If the therapeutic indication of an unregistered medicinal product differs from the indication specified in the summary of product characteristics, it shall not be considered to be a medicinal product for compassionate use (the use of such medicinal product shall be considered to be "off-label use"). [27 July 2010; 17 March 2020] 94.1 The condition referred to in Sub-paragraph 94.2 of this Regulation shall not be applicable to the medicinal products: 94.1 1. not subject to the centralised registration procedure pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council; 94.1 2. for the distribution of which the authorisation referred to in Sub-paragraph 94.1 of this Regulation has been issued; 94.1 3. registered under the centralised registration procedure pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council, but the conditions for the use of such medicinal products and the target group differ from the information specified in the registration documentation of the registered medicinal products. [27 July 2010] 94.2 The authorisation referred to in Sub-paragraph 94.1 of this Regulation for the distribution of unregistered medicinal products for individually granted medicinal products to a clearly known patient or individual patients (Annex 6), by fulfilling the bona fide request, shall not be necessary to the merchant and performer of economic activity referred to in Paragraph 12.1 of this Regulation which has received the unregistered medicinal products from the wholesaler of medicinal products to which the authorisation referred to in Paragraph 94 of this Regulation has been issued. [27 July 2010; 11 September 2012; 8 October 2013] 94.3 In order to receive the authorisation referred to in Paragraph 94 of this Regulation, the person referred to in Paragraphs 94.4 and 94.5 of this Regulation shall submit a submission (Annex 7) to the State Agency of Medicines to which the following shall be appended: 94.3 1. the request of a pharmacy referred to in Sub-paragraph 72.2 of this Regulation for the acquisition of medicinal products if medicinal products are ordered by a pharmacy, and the request of a medicinal treatment institution, social care institution, veterinary medical care institution, or practising veterinarian for the acquisition of medicinal products if the medicinal products are ordered by a medical treatment institution, social care institution, veterinary medical care institution, or practising veterinarian by appending the relevant opinion of the professional association of doctors or professional panel of veterinarians (if any). The abovementioned request for the acquisition of medicinal products may be omitted, if the medicinal products comply with the list of usable medicinal products or are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment. The requirement shall not apply to the authorisation referred to in Sub-paragraph 94.2 of this Regulation; 94.3 2. the labelling of the medicinal product, package leaflet and translation thereof in the official language, if the medicinal products are intended to be delivered to a medical treatment institution, social care institution, practising veterinarian and institutions engaged in veterinary medical care. The requirement shall not apply, if the medicinal products are intended to a clearly known patient, and shall not apply to the authorisation referred to in Sub-paragraph 94.2 of this Regulation; 94.3 3. in regard to medicinal products for compassionate use: 94.3 3.1. a certification of the authorisation requester on the compliance with the requirement stipulated in Article 83(2) of Regulation (EC) No 726/2004 of the European Parliament and of the Council that the submission for the registration of the particular medicinal products has been submitted to the European Medicines Agency or that the medicinal products are undergoing clinical trials; 94.3 3.2. justification of a medical treatment institution for the use of medicinal products grounded with medicinal or epidemiological data on the conformity of a patient and patient group (a special treatment programme) with Article 83(2) of Regulation No 726/2004 of the European Parliament and of the Council confirming that the medicinal products are intended for a chronically or seriously debilitating disease or a disease which is considered to be life-threatening and a satisfactory result of medical treatment cannot be achieved by treating patients with the medicinal products registered in Latvia under the national registration procedures, mutual recognition registration procedures, and decentralised registration procedure or with the medicinal products registered in the European Union in accordance with the centralised registration procedures. If the submitter of the authorisation confirms the implementation of the requirements referred to in Article 83(2) of Regulation No 726/2004 of the European Parliament and of the Council and indicates a medical treatment institution (name and registration number in the register of medical treatment institutions of the Health Inspectorate) where it is intended to implement the programme referred to in Sub-paragraph 94.3 3.3 of this Regulation, the justification for the use of the relevant medicinal products may be submitted later - until the time of delivery of the relevant medicinal products at the medical treatment institution; 94.3 3.3. the programme for the use of the medicinal products drawn up by the authorisation requester. The description of the abovementioned programme shall contain information on the relevant medicinal products and the use thereof: 94.3 3.3.1. a draft summary of product characteristics (if any), a draft of the medicinal product labelling, and a draft package leaflet in the official language; 94.3 3.3.2. information meant for a patient, including a sample form for the informed consent in accordance with the Law on the Rights of Patients; 94.3 3.3.3. description of the group of patients, including the foreseeable number of patients in Latvia, information concerning whether the relevant medicinal products are used in other countries, the expected duration of use and the quantity of the packaging units; 94.3 3.3.4. the obligations of the medical practitioner and recipient of the authorisation, including supervision of the patient, data accumulation, pharmacovigilance, the procedures by which a medical practitioner shall notify of the adverse effects of the medicinal products, the current and planned clinical trials (also the sponsor, site of the clinical trials in Latvia and abroad shall be indicated); 94.3 3.4. if necessary, a copy of the registration submission and the data and documents appended thereto submitted to the European Medicines Agency; 94.3 3.5. an opinion of the Committee for Medicinal Products for Human Use of the European Medicines Agency (if any) and an opinion of the competent authority of another European Community Member State (if any) on the relevant medicinal products; 94.3 3.6. the name and licence number of the wholesaler of medicinal products which will deliver medicinal products to the medical treatment institution referred to in Sub-paragraph 94.3 3.2 of this Regulation shall also be indicated. [27 July 2010; 8 October 2013; 17 March 2020] 94.4 The wholesaler of medicinal products referred to in Paragraphs 11 and 13 of this Regulation is entitled to submit a submission for the receipt of the authorisation referred to in Sub-paragraph 94.1 of this Regulation. [27 July 2010; 11 September 2012; 2 February 2016] 94.5 The person in whose name it is anticipated to register medicinal products under the centralised registration procedure pursuant to Regulation (EC) No 726/2004 of the European Parliament and of the Council and who has submitted the submission for the registration of the medicinal products to the European Medicines Agency or in whose name the authorisation for the commencement of clinical trials has been issued is entitled to submit the submission for receipt of the authorisation referred to in Sub-paragraph 94.2 of this Regulation. [27 July 2010] 94.6 Prior to granting the authorisation referred to in Sub-paragraph 94.2 of this Regulation to medicinal products for compassionate use: 94.6 1. the State Agency of Medicines shall notify the European Medicines Agency of the possibility of using the relevant medicinal products for compassionate use and may request an opinion on the abovementioned medicinal products from the Committee for Medicinal Products for Human Use of the European Medicines Agency: 94.6 1.1. patients of the target group - a restricted group of inhabitants, including age groups which gain benefit during the medical treatment (in respect of the therapeutic indication provided) and which are identified by the Committee for Medicinal Products for Humans Use of the European Medicines Agency; 94.6 1.2. conditions for the use of medicinal products - recommendations for health care professionals regarding safe and effective designation and use of the medicinal products, comprising the relevant information on the clinical, pharmacological and pharmaceutical properties of the medicinal products and the conditions for the supervision of patients; 94.6 1.3. conditions for the distribution of medicinal products - restrictions on or circumstances for the delivery and use of the medicinal products pursuant to Article 9(4)(b) of Regulation (EC) No 726/2004 of the European Parliament and of the Council (shall not apply to the delivery strategy in Member States, for example, quantity of the product, choice of the Member State); 94.6 2. the relevant manufacturer of the medicinal products (if the submission for registration has not been submitted to the European Medicines Agency) or the authorisation requester (the submission for registration has been submitted to the European Medicines Agency): 94.6 2.1. is entitled to inform the European Medicines Agency, upon its own initiative, of the request for the authorisation submitted to the State Agency of Medicines which is referred to in Sub-paragraph 94.2 of this Regulation; 94.6 2.2. shall inform the State Agency of Medicines of the result of consultations with the European Medicines Agency. [27 July 2010; 17 March 2020] 94.7 The authorisation referred to in: 94.7 1. Sub-paragraph 94.1.1 of this Regulation shall be issued in the name of the authorisation requester; 94.7 2. Sub-paragraph 94.2 of this Regulation shall be issued in the name of the wholesaler of medicinal products which will deliver the medicinal products to a medical treatment institution. [27 July 2010; 17 March 2020] 94.8 In the authorisation referred to in Paragraph 94 of this Regulation: 94.8 1. for the medicinal products other than on the list of usable medicinal products or which are not distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment or for the acquisition of which there is no request of a medical treatment institution or social care institution with an opinion of the professional association of doctors respectively, or for the acquisition of which there is no request of a veterinary medical care institution and practising veterinarian with an opinion of the professional panel of veterinarians respectively, the number of packagings shall be indicated after the distribution of which a new authorisation is required for repeated distribution of medicinal products (for bringing in from third countries). This condition shall apply to the authorisation referred to in Sub-paragraph 94.1.1 of this Regulation; 94.8 2. for the medicinal products which correspond to the list of usable medicinal products or which are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment upon request of a medical treatment institution or social care institution to which an opinion of the professional association of doctors is appended, or upon request of a veterinary medical care institution or practising veterinarian to which an opinion of the professional panel of veterinarians is appended, the number of packagings shall not be indicated and authorisation shall be granted for one year. This condition shall apply to the authorisation referred to in Sub-paragraph 94.1.1 of this Regulation; 94.8 3. for medicinal products for compassionate use the term of validity and the quantity of the packaging units need not be indicated, taking into account the information on the anticipated number of patients, duration of the use of the medicinal products and quantity of the packaging units specified in the description of the programme referred to in Sub-paragraph 94.3 3.3 of this Regulation. This condition shall apply to the authorisation referred to in Sub-paragraph 94.2 of this Regulation. [27 July 2010; 2 February 2016; 17 March 2020] 94.9 Prior to assigning the medicinal products referred to in Sub-paragraph 94.2 of this Regulation to a patient, there shall always be a possibility for considering involving in a clinical trial. A medical practitioner who initiates medical treatment with particular medicinal products shall: 94.9 1. inform the particular patient or another person having the right to agree to the treatment of the patient in accordance with the Law on the Rights of Patients (hereinafter - the informed person) of: 94.9 1.1. the circumstances leading to assigning the particular medicinal products; 94.9 1.2. the medicinal products assigned, and shall provide the characteristics thereof (for example, the anticipated benefit, possible risk); 94.9 1.3. supervision of the patient; 94.9 2. receive a written consent of the patient or informed person to the use of the particular medicinal products in accordance with the sample referred to in Sub-paragraph 94.3 3.3.2 of this Regulation. [27 July 2010] 94.10 A medical practitioner or a medical treatment institution shall prepare the substantiation for the use of medicinal products referred to in Sub-paragraph 94.3 3.2 of this Regulation after having acquainted themselves with the programme for the use of medicinal products referred to in Sub-paragraph 94.3 3.3 of this Regulation which has been drawn up by the authorisation requester and which the medical practitioner or the medical treatment institution shall receive from the person referred to in Paragraph 94.5 of this Regulation. [27 July 2010] 94.11 If the authorisation referred to in Sub-paragraph 94.2 of this Regulation which confirms the development of the programme for the use of medicinal products referred to in Sub-paragraph 94.3 3.3 of this Regulation, the person referred to in Paragraph 94.5 of this Regulation shall ensure: 94.11 1. compliance with the conditions stipulated in Articles 24(1) and 83(8) of Regulation (EC) No 726/2004 of the European Parliament and of the Council; 94.11 2. fulfilment of the conditions referred to in Paragraph 94.12 of this Regulation; 94.11 3. submission of the grounded justification for the use of medicinal products of the medical treatment institution referred to in Sub-paragraph 94.3 3.2 of this Regulation to the State Agency of Medicines before commencement of the distribution to a particular medical treatment institution; 94.11 4. submission of the programme for the use of medicinal products agreed upon with the State Agency of Medicines to a medical treatment institution which is referred to in Sub-paragraph 94.3 3.2 of this Regulation; 94.11 5. once in a quarter, submission of the information on the course of the programme for the use of medicinal products to the State Agency of Medicines, but on planned or early completion of the programme for the use of medicinal products - immediately. [27 July 2010; 17 March 2020] 94.12 Medicinal products for compassionate use shall be distributed free of charge. [27 July 2010] 94.13 The authorisation referred to in Sub-paragraph 94.1 of this Regulation for the distribution of unregistered medicinal products for individually granted medicinal products to a clearly known patient or individual patients (Annex 6), by fulfilling the bona fide request, shall not be necessary to the merchant or performer of economic activity referred to in Sub-paragraph 11.1 of this Regulation which has received the unregistered medicinal products from the wholesaler of medicinal products to which the authorisation referred to in Paragraph 94 of this Regulation has been issued. In such case the norm referred to in Sub-paragraphs 95.2.1 and 95.2.2 of this Regulation is not applied. [8 October 2013; 2 February 2016] 95. The person who has been issued the authorisation referred to in Paragraph 94 of this Regulation for the distribution of unregistered medicinal products shall: 95.1. prior to commencing the distribution of the medicinal products notify the State Agency of Medicines of the batch numbers of the medicinal products to be distributed; 95.2. is entitled to distribute the unregistered medicinal products indicated in the authorisation only to the pharmacies, medical treatment institutions, social care institutions, practising veterinarians and institutions engaged in veterinary medical care according to the information indicated in the authorisation (Annex 6). If the authorisation has been granted to unregistered medicinal products which: 95.2.1. conform to the list of usable medicinal products, the medicinal products indicated in the authorisation shall be distributed only to hospitals and to pharmacies of closed type or of medical treatment institutions and of general or open type; 95.2.2. are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient medical treatment, the medicinal products indicated in the authorisation shall be distributed to pharmacies only; 95.2.3. [17 March 2020]; 95.3. prior to commencing the distribution of the medicinal products notify the State Agency of Medicines of the quantities of medicinal products imported and to be distributed; 95.4. notify without delay of all variations made in the data and documents submitted for the receipt of the authorisation; 95.5. issue medicinal products for compassionate use to a patient free of charge; 95.6. [17 March 2020]. [27 July 2010; 2 February 2016] 95.1 The wholesaler of medicinal products referred to in Paragraphs 11 and 13 of this Regulation which brings in unregistered medicinal products for which the authorisation referred to in Paragraph 86 of this Regulation has been issued, prior to commencing the distribution of medicinal products, shall notify the State Agency of Medicines of the medicinal products to be distributed (name, strength or concentration, form, quantity in one packaging and number of packagings) and the batch numbers of the medicinal products. [3 December 2013; 2 February 2016] 95.2 Medicinal products for compassionate use shall be issued only to a group of patients specified in the programme for the use of medicinal products, taking into account the information indicated in the justification for the use of medicinal products for patients and groups of patients for which the medicinal products are to be used. A medical treatment institution shall ensure the fulfilment of the obligations specified in the programme for the use of medicinal products. [17 March 2020] 96. The person who is referred to in Paragraphs 86, 94.4, and 94.5 of this Regulation shall cover expenses for the expert-examination of a submission and documentation according to the price list of paid services of the State Agency of Medicines. [17 March 2020] 97. The State Agency of Medicines shall send the authorisation for the distribution of unregistered medicinal products to the electronic mail address of the person referred to in Paragraphs 86, 94.4, and 94.5 of this Regulation. If the abovementioned person wants to receive the authorisation in the form of a printed document, it shall be issued within three working days for additional fee according to the price list of paid services provided by the State Agency of Medicines. [17 March 2020] 98. The State Agency of Medicines shall take the decision to cancel the authorisation for the distribution of unregistered medicinal products if: 98.1. distribution of registered medicinal products has been commenced on the market of Latvia; 98.2. the State Agency of Medicines has information related to the safety, quality and efficacy of the medicinal product; 98.3. false information has been provided on the unregistered medicinal products. [27 July 2010; 2 February 2016; 17 March 2020] IX. Distribution of Medicinal Products by Means of Information Society Services, Postal Consignments, and Medicinal Products for Personal Use[17 March 2020] 99. By using information society services in accordance with the Law on Information Society Services it shall be permitted to distribute in retail (to inhabitants) only non-prescription medicinal products, except for the case referred to in Paragraph 991 of this Regulation. [14 December 2021] 99.1 A general (open) type pharmacy (branch thereof) shall be permitted to remotely process orders of a private individual - medicinal products, including prescription medicinal products, and medicinal products and medical devices reimbursed from the funds of the State budget - and delivery thereof to the place of residence of the private individual in conformity with the following requirements: 99.1 1. a pharmacy which is selling medicinal products and medical devices to persons at their place of residence shall, before commencement of the service, inform the State Agency of Medicines of ensuring such service. If a person applies to a pharmacy which does not make delivery of medicinal products or has temporarily discontinued it, the pharmacy shall inform the person of the nearest general (open) type pharmacy where such service is provided; 99.1 2. medicinal products shall be delivered to the place of residence of a person if the person may not acquire them in person at a pharmacy or delegate another person to receive them in the unified electronic information system of the health sector; 99.1 3. a pharmacy (branch thereof) shall accept an order of medicinal products via phone or website, or via electronic mail, or by using other means; 99.1 4. during registration of an order, an employee (a pharmacist or pharmacist's assistant) of a pharmacy (branch thereof) or in the case referred to in Section 42 of the Pharmaceutical Law - a medical practitioner - shall identify the consignee of medicinal products by his or her given name, surname, address of the place of residence, and telephone number and provide remote free of charge information and consultation on the abovementioned medicinal products and use of the medicinal products. If a person orders prescription medicinal products to be delivered, the employee of the pharmacy or the branch thereof shall find out also the personal identity number in order to process the e-prescription in the unified electronic information system of the health sector; 99.1 5. for the medicinal products which are referred to in Article 2 of the Delegated Regulation 2016/161, a pharmacy shall fulfil the obligations specified in Articles 10, 11, 13, 25, 27, 28, 29, and 30 of the abovementioned Regulation, including shall verify the safety requirements and, before delivery of the medicinal products, delete the unique identifier, and also shall verify the packagings of medicinal products also for unregistered medicinal products with safety features and ensure preservation of the quality of the medicinal products during delivery, including in conformity with the requirements laid down in the laws and regulations regarding epidemiological safety measures; 99.1 6. delivery of narcotic or psychotropic medicinal products shall be made only for the medicinal products prescribed on e-prescription and deliveries of such medicinal products shall be made only by an employee (a pharmacist or pharmacist's assistant) of a pharmacy (branch thereof) or in the case referred to in Section 42 of the Pharmaceutical Law - a medical practitioner. Restrictions are not laid down for the delivery of other medicinal products to the place of residence of the person in respect of the person who is making it; 99.1 7. the manager of a pharmacy shall ensure and be responsible for the quality of the delivery service provided and the supervision of all stages of delivery - acceptance of an order, consultations on medicinal products, packaging of the ordered medicinal products, labelling and attaching of a sticker on packagings, delivery and handing out of consignment to the person who made the order. The conditions for the storage and transportation during delivery of medicinal products may not affect the quality of medicinal products. [14 December 2021 / Paragraph shall come into force on 1 December 2022. See Paragraph 171.25] 100. The retail trade distribution of non-prescription medicinal products remotely by using information society services shall be permitted only to a general or open type pharmacy which has fulfilled the following requirements: 100.1. provided the inhabitants with a possibility to reach the pharmacy by means of the web during the working hours of the pharmacy and to receive free of charge information and consultations therefrom on the abovementioned medicinal products for the provision of pharmaceutical care in accordance with the laws and regulations regarding the requirements for the opening and operation of pharmacies; 100.2. in conformity with Paragraph 103 of this Regulation developed the website which is available to the public continuously at any time of day or night, and the information inserted therein conforms to the laws and regulations regarding the advertising of medicinal products; 100.3. received a special authorisation (licence) for the opening of a general or open type pharmacy in annex to which the permitted condition of special activity "distribution of non-prescription medicinal products by means of the web" is indicated; 100.4. notified the Health Inspectorate and the State Agency of Medicines of the date from which it offers to distribute non-prescription medicinal products in retail trade by using information society services, and also the website address intended for such purpose, all the relevant information necessary to identify the relevant website, including the domain name and electronic mail address, and also ensures updating of the abovementioned information. [27 July 2010; 8 October 2013; 17 March 2020; 14 December 2021] 101. If a pharmacy distributes medicinal products in retail by means of the web, the owner of the special authorisation for the opening (operation) of a pharmacy shall ensure the necessary power of the server and the capacity of the communication channel connection of the server. [27 July 2010] 102. A pharmacy which distributes non-prescription medicinal products in retail by using information society services shall, without delay, ensure updating of the information provided on the website. [17 March 2020] 102.1 By using information society services, a pharmacy shall distribute non-prescription medicinal products in retail trade to other European Union or European Economic Area Member States only if the medicinal products are classified as non-prescription medicinal products in the particular country. [17 March 2020] 103. In addition to the requirements which are laid down in the Law on Information Society Services and the laws and regulations regarding information society services, the website referred to in Sub-paragraph 100.2 of this Regulation shall have the following structure and content: 103.1. the provision of the functions of the pharmacy and electronic communication possibilities: 103.1.1. the report of services provided by the pharmacy with explanatory particulars regarding the possibility of receipt of the services; 103.1.2. if a submission on a specific printed form is necessary for the receipt of a service, the availability of the printed forms shall be ensured, and the option to print them from the website (in PDF or other universal formats) or to fill them in directly on the website shall be ensured, as well as explanations regarding the correct filling in of the forms. If forms are being filled in several languages, explanations shall be provided also in the relevant languages; 103.1.3. availability of interactive elements (for example, "Your Question", "Answer of Pharmacy", "Submissions, Complaints", "Answers to Submissions, Complaints"); 103.1.4. a possibility to send a letter (a submission) to the pharmacy by electronic means (for example, using XML); 103.1.5. the electronic mail address shall be indicated and a possibility to send suggestions regarding the website; 103.2. information on the pharmacy: 103.2.1. the name of the pharmacy, address, working hours, telephone and fax number, the number of the special authorisation (licence), the firm name of the special authorisation (licence) holder, address, registration and bank details; 103.2.2. the given name, surname and registration number within the Pharmacists' Society of Latvia of such pharmacist who provides consultations regarding medicinal products; 103.2.3. additional graphic elements - a map of the location of the pharmacy with a reference to the closest public transport and a graphic image of the special authorisation (licence) issued to the pharmacy for opening (operation) of the pharmacy; 103.3. the date of the last update of the information placed on the website; 103.4. a link to the opening page, as well as links to pages of a higher level, if the server of the website has a structure of several levels; 103.5. the contact information of the State Agency of Medicines and the Health Inspectorate and the link to the websites thereof; 103.6. a logo conforming to the requirements of Article 1 of Commission Implementing Regulation (EU) No 699/2014 of 24 June 2014 on the design of the common logo to identify persons offering medicinal products for sale at a distance to the public and the technical, electronic and cryptographic requirements for verification of its authenticity (hereinafter - Implementation Regulation No 699/2014) (hereinafter - the common logo) with the following elements: pictogram, in a white rectangle in the middle (left side) of the logo - a picture of the flag of the Republic of Latvia, and a text; 103.7. a link to the website of the State Agency of Medicines in which the list of the licensed pharmacies referred to in Sub-paragraph 103.1 2 of this Regulation is located, has been appended to the common logo in accordance with the requirements laid down in Articles 3 and 4 of Implementation Regulation No 699/2014; 103.8. it is not permitted to add any text, symbol, logo or other element to the field of the common logo, as well as it is not permitted to colour the logo in colours which are not specified in Annex to Implementation Regulation No 699/2014. The logo may not be deformed or its form and visual elements may not be otherwise changed, it may not be disfigured, rotated, multiplied, a symbol and text may not be added. Another logo, symbol or text may not be added at the zone of logo elements. [27 July 2010; 8 October 2013; 2 February 2016; 17 March 2020] 103.1 The State Agency of Medicines shall publish at least the following information on its website: 103.1 1. information on the laws and regulations which are applicable when distributing non-prescription medicinal products in retail trade with the intermediation of information society services, including the information that classification and delivery conditions of medicinal products may differ among Member States; 103.1 2. a list of the licensed pharmacies which are permitted to distribute non-prescription medicinal products in retail by means of information society services and which offer such service, indicating the website addresses of such persons; 103.1 3. information on the risk of use of such medicinal products which are supplied to inhabitants illegally with the intermediation of information society services; 103.1 4. a link to the website of the European Medicines Agency in which the information referred to in Sub-paragraph 103.1 1 of this Regulation is provided, information on the legal acts of the European Union which are applied to counterfeit medicinal products, and also links to websites of the European Union Member States; 103.1 5. information on the purpose of the use of the common logo; 103.1 6. the list of licensed pharmacies (branches thereof) which are remotely processing orders of medicinal products (also medical devices prescribed on prescriptions) of a private individual and making the delivery thereof to the place of residence of the private individual, indicating the name, address, and contact information of such pharmacies, including the website address, if any. [17 March 2020; 14 December 2021 / Sub-paragraph 103.1 6 shall come into force on 1 January 2022. See Paragraph 171.25] 104. The sender of a postal parcel shall be clearly identifiable. Legal persons which receive or send medicinal products in postal consignments shall have at the disposal thereof: 104.1. the special authorisation (licence) referred to in Paragraph 11 or 13 of this Regulation. If medicinal products are received by a medical treatment institution, a statement on the registration in the Register of Medical Treatment Institutions which has been issued by the Health Inspectorate; 104.2. an authorisation issued for each case by the State Agency of Medicines in accordance with the Law on the Legal Trade of Narcotic and Psychotropic Substances and Medicinal Products, and also Precursors if the substances and medicinal products included in Registers II and III are forwarded and received; 104.3. a special authorisation (licence) for the opening (operating) of a general or open type pharmacy. [27 July 2010; 2 February 2016; 17 March 2020; 4 March 2021] 105. The person who sends medicinal products shall submit to the provider of postal services an instruction regarding the conditions for the dispatch of medicinal products (for example, ensuring a specific temperature regime) and conclude an agreement with the provider of postal services to ensure the implementation of the instruction. 106. A natural person has the right to (does not apply to narcotic and psychotropic medicinal products): 106.1. bring in from foreign countries (for example, in a travellerʼs baggage) or receive by post medicinal products for personal use in accordance with Paragraphs 106.1, 106.2 and 106.3 of this Regulation, if they are in the original packaging in a particular pharmaceutical form, their manufacturer and country of manufacture are identifiable on the packaging and they have a purchase cheque or an equivalent document; 106.2. export medicinal products for personal use or send medicinal products by post to another country if it is permitted by the national law of the country to which medicinal products are delivered. [27 July 2010; 2 February 2016] 106.1 A natural person shall not be permitted: 106.1 1. to bring in narcotic analgesics, new psychoactive substances and active substances from foreign countries for personal use; 106.1 2. to receive in a postal consignment: 106.1 2.1. narcotic analgesics, new psychoactive substances and active substances; 106.1 2.2. anabolic steroids, testosterones, growth hormones or their analogues; 106.1 2.3. [5 July 2016]. [2 February 2016; 5 July 2016] 106.2 In the case referred to in Sub-paragraph 106.1 of this Regulation, the quantity of medicinal products provided for personal use is equivalent: 106.2 1. to 12-month use, if the medicinal products are brought in or received in a postal consignment from a country of the European Economic Area; 106.2 2. to 14-day use, if anabolic steroid, testosterone, growth hormone or their analogue is brought in from a country of the European Economic Area; 106.2 3. to six-month use if the medicinal products are brought in from a third country. [2 February 2016] 106.3 If in the case referred to in Sub-paragraph 106.1 of this Regulation: 106.3 1. the quantity of medicinal products exceeds three units of the packaging of the medicinal product (primary, secondary) for each medicinal product, the use of the medicinal product shall be justified by a written confirmation of the person containing an indication regarding the use of the medicinal product for personal needs and a confirmation that the medicinal products are not narcotic analgesics, new psychoactive substances and active substances, as well as anabolic steroids, testosterones, growth hormones or their analogues (if the medicinal products are received by post) and that the person takes responsibility for the use of the such medicinal products. Bringing in of anabolic steroids, testosterones, growth hormones or their analogues shall be justified by a prescription or a document issued by the medical treatment institution; 106.3 2. the medicinal products have been acquired in sale at a distance, the address and licence number of the pharmacy, the website address of the pharmacy, the licence issuing institution and address of the pharmacy shall be indicated on the packaging of the postal consignment; 106.3 3. the medicinal products received by post from third countries shall be issued for a prescription or a document issued by the medical treatment institution. A certified translation in accordance with the laws and regulations regarding the certification of translations into the official language shall be attached to the documents in a foreign language. [2 February 2016; 5 July 2016] X. Quality Control of Medicinal Products107. Prior to commencing the distribution of the medicinal products referred to in Sub-paragraphs 1.3 and 2.2, Paragraphs 3, 4, and 5, and Sub-paragraph 6.2 of Annex 8 to this Regulation, including parallel imported, parallel distributed, and unregistered medicinal products, they shall be subject to quality control. [17 March 2020] 108. If necessary, the State Agency of Medicines shall organise the examination of samples and is entitled to recognise the test results of an official medicinal product control laboratory of another country of the European Economic Area or, if the medicinal products have been manufactured in Latvia, shall declare it to be in conformity with the approved specifications on the basis of the batch manufacturing protocol and the test results. The examination shall be carried out in accordance with the procedures laid down in the Administrative Procedure Law, but not later than within 60 days after receipt of the samples and documentation. 109. The Health Inspectorate is entitled to withdraw samples of medicinal products for examination, except in the cases referred to in Paragraphs 4 and 5 of Annex 8 to this Regulation when the samples for examination are submitted by the marketing authorisation holder. The marketing authorisation holder is entitled not to submit the samples referred to in Paragraphs 4 and 5 of Annex 8 to this Regulation for testing if: 109.1. an Official Control Authority Batch Release (OCABR) of a country of the European Economic Area for the medicinal products has been issued, certifying the conformity of the product with the specifications of the European Pharmacopoeia Monograph for the relevant product and its registration certificate. The marketing authorisation holder shall submit a copy of the abovementioned certificate together with information on the distribution of medicinal products to the State Agency of Medicines. If a justified request to submit samples for testing has not been received from the State Agency of Medicines within seven working days, distribution of the medicinal products may be commenced; 109.2. it has been permitted by the State Agency of Medicines on the basis of public health considerations, if the medicinal products are not available on the market and there is an immediate risk in relation to the spread of virus-caused illnesses or risk to human health or life. [2 February 2016] 109.1 Prior to commencing the distribution of parallel imported, parallel distributed, and unregistered medicinal products in the cases referred to in Paragraphs 4 and 5 of Annex 8 to this Regulation, a wholesaler shall submit the information to the State Agency of Medicines on the batches and supplier of medicinal products intended for distribution. [17 March 2020] 109.2 A wholesaler is entitled to distribute the medicinal products referred to in Paragraph 109.1 of this Regulation after the State Agency of Medicines has informed the wholesaler that it is allowed to distribute medicinal products on the basis of the certificate issued by the official laboratory for the control of medicinal products of the country of European Economic Area (OCABR) or on the basis of public health considerations because medicinal products are not available on the market and there is immediate risk of the spread of illnesses caused by viruses or risk to human health or life. [17 March 2020] 110. A natural person is entitled to submit medicinal products to the State Agency of Medicines for the performance of quality control. 111. Expenses related to the medicinal product sample testing shall be covered by the person from which the samples have been withdrawn, or, in the case referred to in Paragraph 110 of this Regulation - by a natural person in accordance with the price list of paid services of the State Agency of Medicines. [2 February 2016] 111.1 The expenses for testing referred to in Paragraph 111 of this Regulation shall be covered by the State Agency of Medicines for medicinal products which have been registered: 111.1 1. under the mutual recognition procedure and decentralised procedure if samples for testing are taken or sent within the scope of market surveillance which is co-ordinated by the European Directorate for the Quality of Medicines; 111.1 2. under the national registration procedure which is not a mutual recognition procedure and decentralised procedure, if the samples for testing are taken or sent on the basis of the agreement entered into on 13 October 2011 by and between the State Agency of Medicines and the relevant competent authorities of Estonia and Lithuania for a joint procedure in the testing of medicinal products in the field of supervising the quality of medicinal products for ensuring the supervision and control of medicinal products, including upon request of the relevant competent authorities of Lithuania and Estonia, as well as within the scope of market surveillance which is co-ordinated by the European Directorate for the Quality of Medicines. [11 September 2012] XI. Action in Emergency Situations and Procedures for Recall of Medicinal Products112. The supply of medicinal products is prohibited and the medicinal products shall be recalled from the market if: 112.1. the medicinal products are harmful; 112.2. the medicinal products do not have a therapeutic efficacy; 112.3. the risk-benefit ratio is unfavourable; 112.4. the qualitative and quantitative composition of the medicinal product does not conform with that declared in the registration documentation; 112.5. the control of medicinal products or raw materials thereof and the intermediate stage control of the manufacturing process has not been carried out, or any other requirement or obligation related to the granting of the special authorisation (licence) for the manufacture/import of medicinal products has not been fulfilled. [8 October 2013] 112.1 When taking the decision to suspend the distribution of medicinal products or to recall medicinal products from the market, the Health Inspectorate shall indicate a specific date from which the distribution of medicinal products must be suspended or the medicinal products must be recalled from the market, as well as shall indicate the level of suspension or recall of the medicinal products (for example, wholesale trade of medicinal products, retail trade of medicinal products, users of medicinal products) and the batch numbers of the medicinal products to be recalled, if individual batches are being recalled. [27 July 2010; 2 February 2016] 112.2 The Health Inspectorate may take the decision referred to in Paragraph 112.1 of this Regulation also on potentially counterfeit medicinal products and suspicions of quality defects of medicinal products. [8 October 2013] 113. A prohibition for the supply or recall of medicinal products from the market may be applied to such batches of medicinal products for which there are suspicions of a potential counterfeit or quality defect. [8 October 2013] 113.1 In relation to medicinal products the supply of which is prohibited or which have been recalled from the market in accordance with Paragraphs 112 and 113 of this Regulation, the Health Inspectorate together with the State Agency of Medicines, in emergency circumstances in a transitional period, are entitled to supply the medicinal products to patients who are already being treated with them (shall not apply to counterfeit medicinal products). [8 October 2013; see Paragraph 171.6] 114. The recall of medicinal products may be partial, if the medicinal product batch is withdrawn from selected particular distributors or users. 115. A marketing authorisation holder shall notify the State Agency of Medicines and the Health Inspectorate: 115.1. without delay - of any activity which he or she executes to suspend the distribution of medicinal products, to request the recall of medicinal products from the market, to request the cancellation of registration or not to apply for the re-registration of medicinal products, and of the justifications for the relevant activity. The marketing authorisation holder shall particularly indicate whether the implemented activity is based on any of the criteria referred to in Paragraph 112 of this Regulation when the supplies of medicinal products are prohibited and the medicinal products are recalled from the market, or on the criteria for taking the decision referred to in the laws and regulations regarding the procedures for registering medicinal products in relation to suspending or cancelling registration or re-registration of medicinal products or in relation to amending registration of medicinal products (shall apply to the cases when it is recognised that the medicinal products are harmful, medicinal products do not have therapeutic efficacy, the risk-benefit ratio is unfavourable, the qualitative or quantitative composition of the medicinal product does not conform with that declared in the registration documentation, the data indicated in relation to the registration or re-registration submission are not accurate or complete, or amendments have not been made thereto, the obligations specified for the marketing authorisation holder have not been fulfilled in relation to pharmacovigilance, or the medicinal products have not been manufactured according to the description of the manufacturing method, or control is not performed according to the description of control methods). If the action is based on any of the abovementioned criteria, the marketing authorisation holder shall inform the Health Inspectorate and the State Agency of Medicines also in cases if the activity has been implemented in a third country. The marketing authorisation holder shall also inform the European Medicines Agency if the action is based on any of the abovementioned criteria; 115.2. of each established quality defect of medicinal products in conformity with their class in accordance with Annex 9 to this Regulation if, due to the quality defect, a recall of the medicinal products might follow, and shall indicate the country which received the defective product. [21 October 2008; 8 October 2013; 2 February 2016] 115.1 Paragraph 115 of this Regulation shall be applied in relation to medicinal products registered in Latvia. The marketing authorisation holder shall submit information on the medicinal products registered under the centralised registration procedure to the European Medicines Agency. [2 February 2016] 115.2 The information referred to in Sub-paragraph 115.1 of this Regulation shall be submitted electronically together with an accompanying letter (drawn up in MS Word format) in accordance with Annex 8.1 to this Regulation to which the following information is appended (in MS Excel format according to the template approved by the European Medicines Agency): 115.2 1. the reporter - given name, surname, electronic mail address, telephone number; 115.2 2. the marketing authorisation holder - name, address; 115.2 3. regarding the medicinal product - the registered name of the medicinal product in the country of the European Economic Area, active substance, registration procedure; 115.2 4. the activities implemented with medicinal products - activity of the marketing authorisation holder (intended activity), cause, explanation of the cause of the activity (intended activity); 115.2 5. the country in which activity is implemented - it shall be indicated whether the activity has been implemented in a country of the European Economic Area. If the answer is "yes", the country shall be indicated. It shall be indicated if the activity has been implemented in a third country. If the answer is "yes", the country and the name of the medicinal product in such country shall be indicated; 115.2 6. the foreseeable date beginning from which the medicinal product will not be available on the market due to the intended activity. The foreseeable date beginning from which it is anticipated that the medicinal product will be available again on the market after the activity has taken place; 115.2 7. additional information on the medicinal product - registration number, date of the first registration and re-registration, name of the medicinal product, strength or concentration, form of the medicinal product, route of administration. It shall be indicated whether the medicinal product is currently on the market. If the medicinal product is not currently available on the market, the reason shall be indicated. [2 February 2016] 115.3 If a notification is submitted to the European Medicines Agency, it shall be completed in English. [2 February 2016] 115.4 The website of the State Agency of Medicines shall contain a link to the website of the European Medicines Agency in which a sample notification (in English) to be sent to the competent authorities and the European Medicines Agency which is referred to in Paragraph 115.2 of this Regulation, is published. [2 February 2016] 116. If there are suspicions of potentially counterfeit medicinal products or medicinal products of potentially poor quality, the persons referred to in Paragraphs 11 and 13 of this Regulation, the merchant or performer of economic activity who has been issued a special authorisation (licence) for the opening (operation) of a pharmacy, medical treatment institutions, social care institutions, practising veterinarians, institutions engaged in veterinary medical care or other persons shall notify the Health Inspectorate thereof. The following shall be indicated in the notification: 116.1. the name of the medicinal product, strength or concentration, and form of the medicinal product; 116.2. the manufacturer and the marketing authorisation holder (if any) indicated on the labelling and in the package leaflet of the medicinal product; 116.3. the batch number of the medicinal product; 116.4. the person from which the medicinal product has been acquired. [8 October 2013] 116.1 If there are suspicions that the medicinal product significantly endangers public health, the Health Inspectorate in co-operation with the State Agency of Medicines shall evaluate the situation, immediately send a rapid alert notification to all other European Union Member States and ensure sending of notifications to all existing participants of the supply chain, in conformity with the proceedings and procedures laid down in Chapter XII of this Regulation and the time periods specified in Paragraph 122 of this Regulation. If it is assumed that such medicinal products have reached patients, urgent public notifications shall be provided within 24 hours in accordance with Paragraphs 117 and 118 of this Regulation to patients on the recall of the abovementioned medicinal products. The abovementioned notifications shall contain sufficient information on the potential quality defects or counterfeit of medicinal products and the risk related thereto. [8 October 2013] 116.2 The responsible official referred to in Section 38, Paragraph one and Sections 46.1, 52, and 52.1 of the Pharmaceutical Law shall be responsible for the enforcement of Paragraph 116 of this Regulation. [17 March 2020] 116.3 Notifications on suspicions of a defect or counterfeit in accordance with Paragraph 116 of this Regulation shall be submitted to the Health Inspectorate also by the marketing authorisation holder. [2 February 2016] 116.4 The State Agency of Medicines and other testing laboratories may report to the Health Inspectorate on the results which arise from testing the medicinal products on the market and which require further research. [2 February 2016] 116.5 If the Health Inspectorate, upon receipt of a notification, suspects counterfeit of medicinal products or raw materials, the Health Inspectorate shall inform the law enforcement authorities. [2 February 2016] 116.6 The notification referred to in Paragraph 116 of this Regulation shall also apply to a potential fraud in manufacture, deterioration of the product quality or any other serious quality problem related to medicinal products. [2 February 2016] 117. The Health Inspectorate, where necessary, in co-operation with local governments shall ensure that the report on quality control or on the recall of medicinal products is published in periodicals and other mass media (for example, television and radio). [21 October 2008] 118. In emergency situations when a medicinal product is found to be dangerous to consumers' life or may pose a risk to consumers' health, the Ministry of Health shall, on the basis of the report from the Health Inspectorate and the opinion of the State Operational Medical Commission, take a decision and on the same day shall notify the National Radio and Television Council of the statement to be announced to the public of Latvia. [21 October 2008] 119. If the decision to suspend the distribution of medicinal products and to recall medicinal products from the market has been taken, the relevant marketing authorisation holder or the manufacturer/importer of the medicinal products shall notify the persons to which the medicinal products have been supplied of the taken decision by telephone or fax, or using other means of communication. If the decision is communicated orally, the notification in writing shall be submitted concurrently. The following shall be indicated in the notification: 119.1. the name, strength, form and, where necessary, the batch number of such medicinal products to which the notification on the suspension of medicinal product distribution and the recall of medicinal products from the market applies. The date from which, according to the decision on the suspension of the medicinal product distribution and recall of the medicinal products from the market, the distribution of the medicinal products is to be suspended and the medicinal products are to be recalled from the market; 119.2. the marketing authorisation holder and the manufacturer/importer; 119.3. the reason for the suspension of medicinal product distribution and recalling of medicinal products from the market; 119.4. the class of quality defect (first, second or third) and a description; 119.5. the level of urgency of the relevant recall (time in hours at which the persons to whom the medicinal products have been supplied shall be notified) in accordance with Paragraph 122 of this Regulation; 119.6. whether all the clients are to be notified of the recall of medicinal products or only those who have received the batch to be recalled; 119.7. whether the medicinal products are to be recalled from the retail trade network or from the whole market of medicinal products; 119.8. further actions with these medicinal products; 119.9. the procedures for returning the medicinal products to the supplier or marketing authorisation holder; 119.10. the procedures for the collection and destruction of medicinal products; 119.11. the procedures for covering losses. [27 July 2010] 120. A person who receives the notification referred to in Paragraph 119 or 126 of this Regulation and which is responsible for the suspension of medicinal product distribution and recall of the medicinal products from the market shall: 120.1. register the following information: 120.1.1. the date and time when the notification on the suspension of medicinal product distribution and recall of the medicinal products from the market was received, as well as the mode of receipt (for example, by telephone, by fax or electronic means); 120.1.2. the notification provider (the name, given name, surname, position, telephone number, fax number, electronic mail address); 120.1.3. the name, strength, form, batch number, manufacturer and marketing authorisation holder of such medicinal products to which the notification on the suspension of medicinal product distribution and recall of the medicinal products from the market applies, as well as the class of quality defect of these medicinal products and quality defect description (Annex 9); 120.1.4. on the person (given name, surname, position) responsible for the suspension of the distribution of the medicinal products and recall of the medicinal products from the market; 120.1.5. the date and time for commencing the suspension of the distribution of the medicinal products and recall of the medicinal products from the market. 120.2. ensure the suspension of the distribution of the medicinal products specified in the notification and recall of the medicinal products, notifying the persons to whom the medicinal products have been supplied thereof as soon as possible, but not later than within a day after receipt of the notification; 120.3. inform the notification provider of the recalled medicinal products (Annex 10). [2 February 2016] 121. The persons referred to in Paragraph 119 of this Regulation shall, within 10 days after the recall of medicinal products from the market is completed, submit to the Health Inspectorate a report on the recall of medicinal products (Annex 10). The Health Inspectorate has the right to reduce the time limit for report submission if the medicinal products are recalled in the case referred to in Paragraph 118 of this Regulation. [21 October 2008] 122. If a quality defect (Annex 9) or counterfeit has been established for medicinal products, the Health Inspectorate in co-operation with the State Agency of Medicines shall evaluate the situation, as well as further action on the basis of public health considerations, as well as shall send the rapid alert notification (Annex 12) to the relevant distributors of medicinal products electronically, and, if necessary, inform them also orally (by telephone): 122.1. as soon as possible, but not later than within 12 hours, if a first class quality defect (Paragraph 1 of Annex 9) is established for the medicinal products or counterfeit medicinal products are established; 122.2. within 24 hours if a second class quality defect (Paragraph 2 of Annex 9) is established for the medicinal products; 122.3. within 48 hours if a third class quality defect (Paragraph 3 of Annex 9) is established for the medicinal products. In such case the requirement for the recall of medicinal products need not be included in the notification. [2 February 2016] 123. If a quality defect has been established for medicinal products exported and supplied to a country of the European Economic Area, the marketing authorisation holder and the wholesaler of medicinal products shall, within four hours after establishing the fact, notify the consignee of the medicinal products in foreign countries and the Health Inspectorate in writing of the product defect. [21 October 2008; 3 December 2013] 124. The wholesaler of medicinal products, a pharmacy, a medical treatment institution, and a social care institution shall appoint a contact person who may be contacted at any time of the day on issues regarding defects and recall of medicinal products. If variations to data have been introduced, they shall be notified without delay, but not later than within three days after introducing the variations. Data on the contact person, indicating the given name, surname, telephone number and electronic mail address, shall be provided: 124.1. by the wholesaler of medicinal products and the pharmacy to the State Agency of Medicines, indicating them in the submission for receipt of the relevant special authorisation (licence) which is referred to in the laws and regulations regarding the procedures for the licensing of pharmaceutical activity. The contact person for the manufacturer of medicinal products may be the responsible official referred to in Section 52 of the Pharmaceutical Law, for the importer of medicinal products - the responsible official referred to in Section 52.1 of the Pharmaceutical Law, in the medicinal product wholesale facility - the responsible official referred to in Section 46.1 of the Pharmaceutical Law, but in the pharmacy - the manager of the pharmacy or the deputy manager of the pharmacy; 124.2. by the medical treatment institution and social care institution by registering in the register of medical treatment institutions or the register of social care institutions in accordance with the laws and regulations regarding the registration of medical treatment institutions or the laws and regulations regarding the registration of social care institutions. [2 February 2016] XII. Duties of Competent Authority and Procedures for Rapid Alert125. Having received the notification referred to in Paragraphs 115 and 116 of this Regulation or the rapid alert notification referred to in this Chapter, the Health Inspectorate shall submit it to the State Agency of Medicines. The State Agency of Medicines shall evaluate whether the defect indicated in the notification is related to the side effects of the medicinal products. The Health Inspectorate together with the State Agency of Medicines and, if the medicinal products are intended to be used by animals - with the Food and Veterinary Service, shall organise the assessment of the notification in respect of the type, amount and urgency of the risk. The following information shall be specified in the assessment: 125.1. if the defect actually exists, what risk there is to human health and, if the medicinal products are intended to be used by animals, to the health of an animal: 125.1.1. the risk to patients and to patients of particular risk groups; 125.1.2. the risk of receiving inappropriate medical treatment; 125.1.3. the risk from inappropriate dosage (the therapeutic index shall be considered); 125.1.4. the immediate risk and long-term risk; 125.2. the possibility that the defect actually exists and occurs in the medicinal products supplied by the manufacturer of the medicinal products; 125.3. the risk to distort national programmes that are designated for the restriction of diseases caused by various viruses if a suspicions of a vaccine defect arise (mutual contamination by virus); 125.4. whether the potential threat to human health and, if the medicinal products are intended to be used by animals, to the health of an animal, is such that emergency measures need to be taken. When negotiating with the manufacturer of the medicinal products, considerations regarding the following shall be evaluated and included in the notification: 125.4.1. other related notifications; 125.4.2. batch distribution (for example, distributed to known inpatient medical treatment institutions, widely distributed in the wholesale network); 125.4.3. the date of the first distribution and the date of the last distribution; 125.4.4. the remaining stock of the medicinal products located at the manufacturer of the medicinal products; 125.4.5. the possibility that other batches of the medicinal products may have the same defect, and that they will be distributed; 125.5. the characterisation of the situation if no other stocks of the medicinal products and alternative products are available, as well as the clinic effect caused by interruption in supply; 125.6. whether further activities and assessments for other batches of the same product or other products are needed, and whether further investigation of the problem is necessary; 125.7. a temporary or permanent prohibition to sell the remaining stocks; 125.8. the necessity for good manufacturing practice inspection in order to prevent the recurrence of a similar situation; 125.9. whether the notification of a medicinal product recall shall be announced to all or specific medical care professionals or the notification shall be broadcasted on television, radio and in other mass media, and whether information shall be publicized on the websites of the competent authorities, press publications or elsewhere. [21 October 2008; 27 July 2010] 126. The Health Inspectorate shall examine each case individually and in co-operation with the State Agency of Medicines shall evaluate the seriousness of the defect, its possible harm to the consumer, the parties involved and the environment, as well as the possible distribution of the defective batch. On the basis of the assessment referred to in Paragraph 125 of this Regulation, the Health Inspectorate shall draw up: 126.1. a rapid alert notification on a quality defect/recall (Annex 12); 126.2. a notification on follow-up activities and activities which are not urgent, as well as provide information about the quality defects (Annex 13). [21 October 2008] 127. The rapid alert procedure is the transmission of information by rapid alert means on the recall of medicinal products if they have a quality defect, they are counterfeit medicinal products and emergency activity is needed to protect the health of human beings and animals. [2 February 2016] 128. Transmission of information shall take place between the competent authorities which are responsible for the medicinal products for human use and for veterinary medicinal products in the countries of the European Economic Area, the countries which are preparing for joining the European Union, and the countries with which the European Community has entered into a mutual recognition contract, the institutions which participate in the Pharmaceutical Inspection Cooperation Scheme, as well as between the European Commission and international organisations - the European Council, the European Directorate for the Quality of Medicines and the World Health Organisation. The rapid alert procedure shall be used: 128.1. to notify the competent authorities of the presence of potential counterfeit in a legal distribution network or of products which are related to fraudulent activities in manufacture, packaging, distribution or advertising, and also of products which contain counterfeit raw materials; 128.2. when transmitting other information, for example, warnings regarding the use and recall of medicinal products due to safety considerations, or notifying of further activities; 128.3. to notify of quality defects, counterfeit or active substances or trial medicinal products obtained by means of fraud if the relevant competent authority deems it essential. [2 February 2016] 129. The Health Inspectorate shall ensure the implementation of the rapid alert procedure at any time of day or night. [2 February 2016] 130. Within the scope of the rapid alert procedure, the Health Inspectorate shall, through the rapid alert system, send such information electronically the urgency and seriousness of which precludes any delay. It shall be a rapid alert notification on: 130.1. a first class defect (Paragraph 1 of Annex 9) and counterfeit which is sent to all contact persons in the list of the rapid alert notification list of the European Medicines Agency regardless of whether the particular batch of medicinal products has or has not been exported to such country. If it is necessary to warn the authorities of countries in different time zones, they shall be contacted by telephone. The notifications shall be sent in accordance with the requirements referred to in Sub-paragraph 122.1 of this Regulation; 130.2. a second class defect which may cause disease or incorrect medical treatment (Paragraph 2 of Annex 9), which is sent to all contact persons in the list of the rapid alert notification list of the European Medicines Agency. If there is information on specific countries in which the medicinal products have been distributed, the notification shall be sent only to the contact persons of the relevant countries. If possible, the notification shall be sent in accordance with the requirement referred to in Sub-paragraph 122.2 of this Regulation, but not later than within 24 hours from provision of the notification in Latvia; 130.3. a third class defect (Paragraph 3 of Annex 9) is not usually sent. [2 February 2016] 131. The rapid alert procedure shall be used also in notifying the relevant authorities of a recall of products or determination of a prohibition to sell the medicinal products, in case of suspending or cancelling a manufacture or wholesale trade licence. [2 February 2016] 132. The rapid alert notification shall be sent to the institutions referred to in Paragraph 128 of this Regulation and to the State Agency of Medicines in English. The list of addressees shall be appended to the notification, and the notification shall be sent in the form of an electronic document to the electronic mail address of the particular institution. [2 February 2016] 133. The Health Inspectorate shall: 133.1. draw up the following internal legal acts: 133.1.1. the procedures by which a rapid alert notification is received; 133.1.2. the procedures by which the rapid alert notification is evaluated; 133.1.3. the procedures by which the rapid alert notification is issued and sent: 133.1.3.1. to persons who are included in the rapid alert list of the European Medicines Agency; 133.1.3.2. to the third countries with which Latvia has concluded a bilateral co-operation contract in the field of health care (to the competent authorities); 133.1.3.3. to distributors and consumers in the Republic of Latvia; 133.1.4. the procedures by which the rapid alert notification on follow-up activities is issued and by which non-urgent information on quality defect is distributed (Annex 13); 133.2. issue an order on the official who is responsible for the transmission of the rapid alert notification, and shall notify the European Medicines Agency thereof, indicating the given name, surname of the official and means of communication during and outside working hours, as well as notify the European Medicines Agency of variations in these data; 133.3. support the marketing authorisation holder in the process of medicinal product recall and shall monitor the effectiveness of the process of medicinal product recall; 133.4. ensure that information on the recall of medicinal products is being notified to the persons referred to in Paragraph 128 of this Regulation as soon as possible, if the quality defect causes a serious risk to public health; 133.5. monitor the conduct of the implementation of the medicinal product recall process; 133.6. investigate the circumstances that led to the distribution of the defective product and ensure that any necessary corrective action is taken by the manufacturer of the medicinal products and, where necessary, also by the marketing authorisation holder; 133.7. perform those follow-up activities which are not urgent, and provide information on the quality defect in accordance with Annex 13 to this Regulation. [21 October 2008] 134. The Health Inspectorate shall examine each case individually and in co-operation with the State Agency of Medicines shall assess the seriousness of the defect, its possible harm to the consumer, parties involved and the environment, and the possible distribution of the defective batch. [21 October 2008] 135. If the defective medicinal products (including parallel imported medicinal products and parallel distributed medicinal products) are first identified in Latvia, the Health Inspectorate together with the State Agency of Medicines shall: 135.1. examine the quality defect and issue an order on rapid alert notification (for medicinal products which have been registered under the national registration procedure); 135.2. lead the examination of the quality defect and issue an order on rapid alert notification (for medicinal products which have been registered under the centralised registration procedure, or medicinal products which have been registered both under the centralised and national registration procedure). The notification shall include recommended activities for all involved competent authorities. [21 October 2008] 136. The Health Inspectorate (in co-operation with the State Agency of Medicines) may come to an agreement with the European Medicines Agency and the representative of the Committee for Medicinal Products of the European Medicines Agency who is the rapporteur on medicinal products in the Committee for Medicinal Products on the actions to be taken. [21 October 2008] 137. The rapid alert notification shall include a description of the different packaging of the medicinal product in which the product may be packed (if information has been received from the European Medicines Agency). 138. If medicinal products have been re-packaged in parallel distribution, but the defect is not related to re-packaging and has resulted from the manufacture of the medicinal products, a description of the different packages in which the product is distributed (if information from the European Medicines Agency is available) shall be indicated in the rapid alert notification. 139. The countries and authorities referred to in Sub-paragraph 128.1.2 of this Regulation shall, within the scope of the Rapid Alert System, be also notified of the fact of medicinal product distribution by sending information om: 139.1. possible counterfeit medicinal products; 139.2. medicinal products resulting from fraud in manufacture, packaging, distribution or promotion; 139.3. a product containing counterfeit raw materials. 140. The Health Inspectorate shall establish and maintain a system that would ensure receipt of a relevant notification in case suspicions of defective products arise, and draw up, maintain and update the list of medicinal products to be recalled, in accordance with the European Commission's Compilation of Community Procedures on Inspections and Exchange of Information. [21 October 2008] 141. The Health Inspectorate shall submit the decision to suspend the distribution of medicinal products and to recall medicinal products, on the date of issue thereof, to the Food and Veterinary Service, the State Agency of Medicines and the Ministry of Health. The Health Inspectorate shall ensure and supervise the enforcement of the decision, including the follow-up activities. [21 October 2008; 22 September 2009] 142. If medicinal products are being withdrawn from trade, the marketing authorisation holder or the manufacturer of medicinal products shall compensate the losses caused to consumers of the medicinal products, and also the expenses arisen to persons who purchased the medicinal products in relation to the collection, transportation to the place of destruction, and destruction of such medicinal products. [27 July 2010] 143. If poor-quality medicinal products have been found and they are withdrawn from trade, the person due to whose fault the medicinal products lost their quality shall cover the losses and expenses referred to in Paragraph 142 of this Regulation. 144. Medicinal products unusable for distribution shall be destroyed in accordance with the laws and regulations governing the circulation of hazardous waste. XIII. Supervision145. The Health Inspectorate shall: 145.1. monitor and check the distribution of parallel distributed medicinal products and the conformity thereof with the medicinal product marketing authorisation of the European Union and amendments made thereto; 145.2. ensure, according to the competence, the implementation of the conditions or restrictions of such European Commission decision which has been taken in accordance with Regulation No 726/2004 of the European Parliament and of the Council and the opinion of the Scientific Committee (referring to recommended conditions or restrictions with regard to the safe and effective use of the medicinal product as specified in Article 9(4)(c) of Regulation No 726/2004 of the European Parliament and of the Council); 145.3. notify the Food and Veterinary Service of the poor-quality and counterfeit medicinal products found in third countries; 145.4. notify the World Health Organisation of the activity commenced (suspension of the distribution of medicinal products or recall of medicinal products from the market) in relation to the poor-quality or counterfeit medicinal products detected, and also of suspicions of those which may affect public health protection in third countries. A copy of the notification shall be submitted to the European Medicines Agency; 145.5. be regarded to be the competent authority referred to in Article 1(f), Article 12(d), Articles 15 and 18, Article 22(d), Article 24, Article 25(4)(b), Article 30, Article 31(2), Article 32(4), Article 35(1)(e) and (i)(ii), Article 36(i), (j), and (m), Article 37(a), (d), (e), (f), and (g), and Articles 39 and 46 and Annexes III and IV to Delegated Regulation No 2016/161; 145.6. establish artificial non-availability of medicinal products; 145.7. control customs warehouses and temporary storage locations of medicinal products. It shall inspect customs warehouses prior to the issuing of a special authorisation (licence) or in relation to the re-registration of a special authorisation (licence) in order to assess the conformity of premises, equipment, and devices, and also the conformity of the staff with Sub-paragraphs 12.4 and 12.7 of this Regulation. [21 October 2008; 22 September 2009; 27 July 2010; 2 February 2016; 15 January 2019; 17 March 2020; 14 December 2021] 146. The State Agency of Medicines: 146.1. shall fulfil the obligation of the competent supervisory authority referred to in Articles 18 and 19 of Regulation No 726/2004 of the European Parliament and of the Council regarding the medicinal products which in accordance with the abovementioned Regulation have been registered under the centralised registration procedure (including in pharmacovigilance) in accordance with Regulation No 726/2004 of the European Parliament and of the Council; 146.2. shall ensure, according to the competence, the implementation of the conditions or restrictions of such European Commission decision which has been taken in accordance with Regulation No 726/2004 of the European Parliament and of the Council and the opinion of the Scientific Committee (abovementioned recommended conditions or restrictions with regard to the safe and effective use of the medicinal product as specified in Article 9(4)(c) of Regulation No 726/2004 of the European Parliament and of the Council); 146.3. shall notify the Food and Veterinary Service: 146.3.1. of the decision of the State Agency of Medicines that is taken in accordance with the laws and regulations regarding the procedures for registering medicinal products on the determination of the conformity of the product with the definition of the medicinal product specified in the Pharmaceutical Law and subjecting the product to registration with the State Agency of Medicines, as well as indicate a deadline by which the decision must be enforced; 146.3.2. regarding cancelled authorisations for the distribution of unregistered medicinal products; 146.4. shall ensure analysis of medicinal product consumption, summarisation and publication of the results; 146.5. is entitled to issue the authorisation referred to Paragraphs 34, 86 and 94 of this Regulation, if a medicinal product, registered in Latvia and included in the Medicinal Product Register of Latvia, cannot be used for the treatment of a patient or a specific disease, or for the performance of a treatment manipulation because the medicinal product is not available to the consumer on the market. In such case: 146.5.1. Paragraph 86 of this Regulation shall be applied after commencing the distribution of such medicinal products; 146.5.2. the authorisation referred to in Paragraph 34 of this Regulation shall be received within one year after commencing the distribution of such medicinal products; 146.5.3. the requirement for the submission of the pharmacy and medical treatment institution request referred to in Paragraph 94 of this Regulation shall not be applied to obtaining the authorisation; 146.6. shall notify the Health Inspectorate of the batches of medicinal products which are not safe and of good quality or may be hazardous to human health or life not later than within one day after establishing the fact; 146.6.1 shall carry out conformity assessment inspections, except for at the customs warehouses, prior to the issuance of a special authorisation (licence) or in relation to the re-registration of a special authorisation (licence) in order to assess the conformity of the relevant premises, equipment, devices, staff, and documents with the requirements for good distribution practice of medicinal products laid down in this Regulation and the conditions of special activity. A report on inspection (Annex 13.1) shall be prepared after the inspection; 146.7. shall carry out inspections on the conformity of the distribution of medicinal products with good distribution practice after the issuance of the special authorisation (licence) referred to in Paragraph 11 of this Regulation at least once in five years and, where necessary, shall carry out inspections which have not been notified. If unannounced inspections are not carried out, an agreement shall be reached with the wholesaler of medicinal products and the person who brokers medicinal products on the time when an inspection is to be commenced, and shall notify the wholesaler of medicinal products and the person who brokers medicinal products thereof in writing. If necessary, the State Agency of Medicines has the right to request the Official Control Authority Batch Release of a country of the European Economic Area or a laboratory of the State Agency of Medicines to perform a sample test; 146.8. may carry out the inspections of wholesalers of medicinal products referred to in Sub-paragraph 146.7 of this Regulation upon request of a European Union Member State, the European Commission or the European Medicines Agency also in the third countries. The inspections may also be carried out in the premises of such persons who broker medicinal products; 146.9. after each inspection referred to in Sub-paragraphs 146.7 and 146.8 of this Regulation, shall prepare a control report in accordance with Annex 13.1 to this Regulation. It shall be indicated in the control report whether the relevant wholesaler of medicinal products and the person who brokers medicinal products meet the requirements of good distribution practice. The State Agency of Medicines shall, within three working days after preparation of the control report, send it to the inspected person in the form of an electronic document to his or her electronic mail address or - upon request - in the form of a printed document, and shall provide a possibility to submit comments. If necessary, the control report shall be sent to the institution which requested the performance of the inspection; 146.10. shall issue a certificate of good distribution practice conformity of medicinal products which conforms to the sample of the good distribution practice certificate of the European Union (Annex 14) if the activity of the inspected person conforms to the requirements of good distribution practice. The authorisation shall be issued in the form of an electronic document by sending it to the electronic mail address of the inspected person within three working days after the relevant wholesaler of medicinal products has paid the specified fee for the assessment of the documents according to the price list of paid services of the State Agency of Medicines. If conformity assessment of good distribution practice is related to a trip, the wholesaler of medicinal products shall cover the travel (transport) expenses of the State Agency of Medicines to the object to be inspected and back, hotel (accommodation) expenses, health insurance expenses, and daily allowance in accordance with the laws and regulations regarding the procedures for reimbursing the expenses related to official travels and work trips of employees. The certificate in the form of a printed document shall be issued within three working days after receipt of a request and for an additional charge for such service according to the price list of paid services of the State Agency of Medicines; 146.11. shall enter information on the issued certificates of good manufacturing practice in the European Union database on manufacturing and import authorisations and good manufacturing practice certificates (hereinafter - the Eudra GMDP database) within three working days after taking the decision to issue the certificate; 146.12. if the result of the inspection referred to in Sub-paragraph 146.7 of this Regulation shows that the wholesaler of medicinal products does not conform to the requirements laid down in the Pharmaceutical Law and this Regulation and the requirement of good distribution practice is not conformed to in the activity thereof, shall enter information in the Eudra GMDP database within three working days after taking of the decision; 146.13. is entitled to issue information to the requester on the wholesale data of exported medicinal products, including parallel imported and parallel distributed medicinal products, (excluding veterinary medicinal products) (excluding references to specific pharmacies, medicinal product wholesale facilities, manufacturers and importers of medicinal products and without indicating other identifying data) which is necessary for analysing the access to medicinal products, in the following division: 146.13.1. a standard report on sales data of medicinal products (quarter in division by months, six months, year) - shall include the anatomically-therapeutic chemical classification number (ATC code), the international nonproprietary name (INN), the form, strength, or concentration, the number in the packaging, the number of packagings sold, the turnover in EUR; 146.13.2. an expanded report on sales data of medicinal products (quarter in division by months, six months, year) - shall include the information included in the standard report and the consignee group of medicinal products in accordance with Sub-paragraph 18.4 of this Regulation or belonging of the medicinal products to a classification group; 146.13.3. a full report on sales data of medicinal products (quarter in division by months, six months, year) - shall include the information included in the standard report, the consignee group of medicinal products in accordance with Sub-paragraph 18.4 of this Regulation and belonging of the medicinal products to a classification group; 146.13.4. an individual report on sales data of medicinal products which is issued only to the marketing authorisation holder for his or her registered medicinal products - shall include the international nonproprietary name (INN), the registration number of medicinal products, the anatomically-therapeutic chemical classification number (ATC code), the form, strength or concentration of medicinal products, the number in the packaging, the number of packagings sold; 146.14. shall enter into a contract with the requester of information for the service of providing wholesale data of medicinal products, including exported, parallel imported and parallel distributed medicinal products (except for veterinary medicinal products) in the cases referred to in Sub-paragraph 146.13 of this Regulation according to the price list of paid services of the State Agency of Medicines; 146.15. shall ensure the conformity of the website referred to in Paragraph 103.1 of this Regulation with the requirements referred to in Sub-paragraph 103.1 5 of this Regulation and in Articles 2, 3 and 4 of Implementation Regulation No 699/2014; 146.16. shall follow the availability of medicinal products and analyse it, forecast tendencies and public health risks; 146.17. shall post the data received on the distribution of particular medicinal products which is referred to in Paragraph 60.2 and Sub-paragraphs 153.3.1 and 153.3.2 of this Regulation without delay on the website of the State Agency of Medicines, indicating the date of commencing or discontinuing distribution of the relevant medicinal products, as well as on the fact of the availability of medicinal products; 146.18. is entitled to request data from the wholesalers of medicinal products and pharmacies on the remaining stock of the particular medicinal product; 146.19. shall be regarded to be the competent authority referred to in Article 15, Article 31(2), Article 32(4), Article 35(1)(e) and (i)(ii), Article 37(a), (e), (f), and (g), and Articles 39, 43, and 44 of Delegated Regulation No 2016/161. [21 October 2008; 22 September 2009; 27 July 2010; 8 October 2013; 2 February 2016; 20 December 2016; 15 January 2019; 4 March 2021; 14 December 2021] 146.1 The State Agency of Medicines shall authorise officials for carrying out the inspection referred to in Sub-paragraphs 146.7 and 146.8 of this Regulation who are entitled: 146.1 1. to inspect the wholesalers of medicinal products and the persons brokering medicinal products; 146.1 2. to take samples in order to perform independent testing at the Official Control Authority Batch Release of a country of the European Economic Area or a laboratory of the State Agency of Medicines. [8 October 2013] 147. If in the case referred to in Paragraph 43 or Paragraphs 1, 2 and 3 of Annex 8 to this Regulation testing of medicinal products is performed by another testing laboratory which is accredited by the State Agency "Latvian National Accreditation Bureau" in conformity with the LVS EN ISO/IEC 17025:2005 standard "General Requirements for the Competence of Testing and Calibration Laboratories" regarding which the Ministry of Economics has published a notification in the official gazette Latvijas Vēstnesis, it has the obligation to notify the State Agency of Medicines of the test results without delay (on the day of establishing the fact). 148. The Health Inspectorate shall place the following information on the recalled medicinal products on the website of the Health Inspectorate: 148.1. the name, strength, form, batch number of the medicinal product, firm name and country of manufacture of the medicinal product, but regarding registered medicinal products - also the registration number; 148.2. the reason of recall and a description of the medicinal product defect. [21 October 2008; 27 July 2010; 8 October 2013] 149. The State Agency of Medicines shall ensure the following information on the website of the State Agency of Medicines: 149.1. the labelling and approved package leaflets for parallel imported medicinal products. If parallel imported medicinal products differ from registered medicinal products, the difference shall be indicated; 149.2. the parallel importers and parallel imported medicinal products. The information shall be prepared in accordance with the laws and regulations regarding the procedures for registering medicinal products which specify the information on parallel imported medicinal products to be published in the Medicinal Product Register of Latvia on the website of the State Agency of Medicines; 149.3. the parallel distributors and the medicinal products for parallel distribution in Latvia according to the information published on the website of the European Medicines Agency; 149.4. [17 March 2020]; 149.5. the website addresses of the medicinal product wholesale facilities and manufacturers of the medicinal products; 149.6. a sample of the submission in Latvian for the notification referred to in Paragraph 63 of this Regulation of parallel distribution of the centrally registered medicinal products; 149.7. the unregistered medicinal products for the distribution of which the authorisation referred to in Paragraph 86 of this Regulation for the distribution of unregistered medicinal products has been issued. The abovementioned medicinal products shall be included in the Medicinal Product Register of Latvia for the term of validity of the authorisation, and the following shall be indicated: 149.7.1. the name of the medicinal products, strength or concentration and the form of the medicinal products; 149.7.2. international name of the active ingredient; 149.7.3. the name, address of the marketing authorisation holder (or marketing authorisation requester) and the name of the country of the European Economic Area which has granted the marketing authorisation or in which a request for the marketing authorisation has been submitted; 149.7.4. the name of the manufacturer of the medicinal products and the name of the country of manufacture; 149.7.5. the code of the anatomical therapeutic chemical classification and defined daily dose; 149.7.6. the identification number of the medicinal products assigned by the State Agency of Medicines; 149.7.7. the designation of the classification group for prescription medicinal products. The designation shall not be indicated for medicinal products not subject to medical prescription; 149.8. the wholesaler (name, licence number, address) of medicinal products which distributes unregistered medicinal products for which the authorisation referred to in Paragraph 86 of this Regulation has been issued; 149.9. the licensed pharmacies, medicinal product wholesale facilities, manufacturers of medicinal products and importers of medicinal products (licence number, name, registration number, legal address, and address of the site of pharmaceutical activity, contact information and types of special activity). In addition at least the manager of the pharmacy, the term of validity of the licence and the conditions of special activity shall be indicated for pharmacies. At least the responsible official shall be additionally indicated for medicinal product wholesale facilities. In addition at least the qualified person and the date of granting the licence shall be indicated for manufacturers of medicinal products and importers of medicinal products; 149.10. the data on the authorisations issued in accordance with Section 48, Paragraph one of the Pharmaceutical Law: 149.10.1. the date and number of the decision to permit the acquisition of medicinal products; 149.10.2. the firm name (for a natural person − the given name, surname), registration number, and address of the place of activity of the submitter; 149.10.3. the name, strength, and pharmaceutical form for the medicinal products which are permitted to be acquired; 149.10.4. the given name, surname, and telephone number of a contact person; 149.11. the list with unregistered medicinal products for the distribution of which the authorisation for the distribution of unregistered medicinal products referred to in Sub-paragraph 94.8 2 of this Regulation has been issued and which have the request of a medical treatment institution, social care institution, practising veterinarian, or veterinary medical care institution for the acquisition of medicinal products to which the relevant opinion of the professional association of doctors or professional panel of veterinarians has been appended, indicating the information referred to in Sub-paragraphs 149.7.1, 149.7.2, 149.7.4, 149.7.5, and 149.7.7 of this Regulation; 149.12. the wholesaler of medicinal products (the name, licence number, address) which is distributing the unregistered medicinal products referred to in Sub-paragraph 149.11 of this Regulation on the basis of the authorisation issued by the State Agency of Medicines for the distribution of unregistered medicinal products referred to in Sub-paragraph 94.1 of this Regulation; 149.13. the indication in the Medicinal Product Register of Latvia whether the relevant medicinal product is available. [27 July 2010; 11 September 2012; 3 December 2013; 17 March 2020] 149.1 In addition to the request referred to in Sub-paragraph 149.13 of this Regulation, the State Agency of Medicines shall indicate on its website at which medicinal product wholesale facility the relevant medicinal products are available, and also the information on the remaining total stocks of the particular medicinal products intended for the market of Latvia. [17 March 2020 / Paragraph shall come into force on 1 April 2020. See Paragraph 171.13] 150. The State Agency of Medicines shall fulfil all the necessary procedures to ensure that officials and employees who are responsible and are involved in the taking of decisions in relation to the issuance of the authorisations specified in this Regulation, as well as developers (rapporteurs) of reports and experts, are not financially or otherwise interested in the field of pharmacy and their impartiality is not influenced. The abovementioned persons, in entering into a contract with the State Agency of Medicines for the performance of the particular work, shall submit to the State Agency of Medicines the annual declaration of financial interest. The declaration shall be appended to the contract. 151. The Health Inspectorate and the State Agency of Medicines: 151.1. shall not disclose information related to commercial secrets of the medicinal product distributor which has become known to them in the course of enforcing this Regulation; 151.2. shall, according to the competence, ensure prompt mutual information exchange and information exchange with other authorities, as well as, in order to prevent direction of medicinal products to illegal circulation, provide information to the law enforcement authorities and the Ministry of Health on the facts that have come to their knowledge; 151.3. shall determine the procedures for receiving and processing the notifications referred to in Paragraphs 115 and 116 of this Regulation in relation to the medicinal products which are suspected of a potential counterfeit or quality defect, and the procedures for recalling batches of medicinal products both during the working hours and outside working hours. [21 October 2008; 8 October 2013] 152. In order to ensure prompt exchange of information in accordance with the requirements of this Regulation, the medicinal product manufacturer, medicinal product wholesale facility, pharmacy, medical treatment institution and social care institution shall have: 152.1. telecommunications; 152.2. a fax machine (that does not apply to the pharmacy, medical treatment institution (the practice of a physician), as well as social care institution, in which the receipt and sending of information is ensured by other technical means); 152.3. a computer with the necessary software (that does not apply to the medical treatment institution and social care institution, in which the receipt and sending of information is ensured by other technical means). 153. The marketing authorisation holder shall: 153.1. be responsible for the advancement of medicinal products on the market, including planning the registration and re-registration process and time limits so that the stocks of medicinal products referred to in Paragraph 78.1 of this Regulation would not increase. The marketing authorisation holder may appoint a person who is a local representative of the marketing authorisation holder in the country of the European Economic Area who represents him or her in the relevant Member State (hereinafter - the representative); 153.2. [17 March 2020]; 153.3. after the State Agency of Medicines has taken the decision to register medicinal products or in relation to medicinal products registered under the centralised registration procedure - after entering into effect of the relevant decision of the European Commission, shall submit a notification to the State Agency of Medicines in writing: 153.3.1. for the actual date of commencing the distribution (sale) of medicinal products in Latvia, indicating for the medicinal products registered in Latvia the product number in the Medicinal Product Register of Latvia. For medicinal products registered under the centralised registration procedure - the European Union number assigned by the European Medicines Agency for each size of the packaging of the form of registered medicinal product; 153.3.2. for the registered medicinal products which are permanently or temporarily not placed on the market of Latvia. Such notification shall be provided at least two months (except for emergency circumstances) before the placing on the market of medicinal products is discontinued. The marketing authorisation holder shall inform the State Agency of Medicines of the reasons for such actions in accordance with Sub-paragraph 115.1 of this Regulation; 153.3.3. for all incidents in relation to the administration of medicinal products and executed or executable activities (in Latvia or another country); 153.4. ensure fulfilment of the requirements laid down in Sub-paragraph 12.8 of this Regulation; 153.5. ensure urgent implementation of the restrictions related to the safety of medicinal products to be distributed which are specified in accordance with the laws and regulations regarding the procedures for registering medicinal products; 153.6. fulfil the obligations laid down for the marketing authorisation holder in accordance with Articles 31, 32, 33, 35, 36, 37, 38, 40, 41, and 42 of Delegated Regulation No 2016/161. The obligation laid down in Article 33 of Delegated Regulation No 2016/161 - to upload the information in the Repository System of the Medicinal Products of Latvia - shall be fulfilled by using the European repository system of medicinal products. [11 September 2012; 8 October 2013; 2 February 2016; 15 January 2019; 17 March 2020; 14 December 2021] 154. Appointing of the representative shall not release the marketing authorisation holder from the obligations which are specified in Paragraph 153 of this Regulation. The representative shall ensure fulfilment of the duties specified in Paragraph 20 and Sub-paragraphs 153.3, 153.4 and 153.5 of this Regulation. 155. If the manufacturer of medicinal products, wholesaler of medicinal products, pharmacy, medical treatment institution, practising veterinarian, institution engaged in veterinary medical care or social care institution terminates activity, it is re-organized or liquidated, the person who is responsible for the re-organisation or liquidation shall transfer for sale the remaining stock of medicinal products to persons who have the right to distribute the medicinal products, or shall destroy the medicinal product stock in accordance with the requirements laid down in Paragraph 144 of this Regulation and submit to the Health Inspectorate information: 155.1. for agreeing upon the use, distribution or disposal of the remaining stock of medicinal products; 155.2. on the administration or distribution of the remaining stock of medicinal products, indicating the legal person to whom medicinal products are distributed or information on the disposal of medicinal products and a certification that the medicinal product stocks are liquidated. [21 October 2008; 27 July 2010; 11 October 2013] 156. Marketing authorisation holders, manufacturers of medicinal products, medical care practitioners and officials and employees of the State Agency of Medicines shall not be subject to civil or administrative liability for any consequences caused by the administration of medicinal products which do not correspond to the registered indications, or consequences caused by the administration of unregistered medicinal products, if such administration has been suggested or requested by the competent authority (medical treatment institution), having considered any possible or already ascertained spread of pathogens, toxins, chemical substances or nuclear radiation. Any of the abovementioned cases may cause harm irrespective of whether the authorisation has been granted in the European Community or medicinal products have been registered in accordance with this Regulation. 156.1 The Health Inspectorate shall notify the State Police and the customs of the distribution of counterfeit and potentially counterfeit medicinal products in Latvia, also on the basis of information which has been obtained in the rapid alert system. [2 February 2016] 156.2 The Health Inspectorate, the State Police, the customs and investigatory institutions shall mutually co-operate in detecting counterfeit medicinal products, also in cases if the medicinal products have been obtained by fraud (upon manufacturing, packaging, distributing or advertising) or using counterfeit raw materials. [2 February 2016] 156.3 The State Police shall inform the State Agency of Medicines and the Health Inspectorate of the illegal use of medicinal products in order to create a specific condition for a person, including illegal sale and administration of medicinal products against the personʼs will for the purpose of violence. [2 February 2016] 156.4 The Health Inspectorate in co-operation with the State Agency of Medicines shall, according to the competence, inform the professional organisation of the field of pharmacy, merchants and public of the danger of distributing counterfeit medicinal products and the measures to be taken in order to restrict their becoming part of illegal distribution chain of medicinal products. [2 February 2016] 156.5 In order to make public information on the preventive measures and the measures taken in the field related to the falsification of medicinal products, the Ministry of Health in co-operation with the Health Inspectorate, the State Agency of Medicines, the State Police and the customs shall organise meetings in which patient and consumer organisations and, if necessary, officials of other competent authorities participate. [2 February 2016] XIV. Closing Provisions157. Cabinet Regulation No. 88 of 27 February 2001, Regulations Regarding the Import, Export and Distribution of Medicinal Products and Requirements for the Opening and Operation of Medicinal Product Wholesale Facilities (Latvijas Vēstnesis, 2001, No. 88, 35, 52; 2003, No. 114; 2004, No. 69), is repealed. 158. [2 February 2016] 159. Until the date when the special authorisation (licence) for the manufacture or import of medicinal products referred to in Sub-paragraph 11.2 of this Regulation is received, such merchants and performers of economic activity are entitled to distribute in wholesale medicinal products manufactured thereby to whom, on the day of coming into force of this Regulation, the special authorisation (licence) has been issued for: 159.1. the opening (operation) of a medicinal product manufacturing undertaking; 159.2. the manufacture (re-packaging and re-dividing up) of medicinal products at a medicinal product wholesale facility; 159.3. the manufacture of medicinal products in a pharmacy. [11 September 2012] 160. Medicinal product wholesale facilities to which, on the day of coming into force of this Regulation, the special authorisation (licence) for the opening of a medicinal product wholesale facility with a condition of special activity - import of medicinal products into Latvia from a country which is not located in the European Economic Area and the authorisation for the import of the relevant medicinal products into the Republic of Latvia from the third countries issued by the State Agency of Medicines has been issued, are entitled to distribute in wholesale the imported medicinal products up to the receipt of the special authorisation (licence) for the manufacture or import of medicinal products. 161. The authorisation for the distribution of unregistered medicinal products which has been issued by the State Agency of Medicines until the day of coming into force of this Regulation shall be valid. After importation of the quantity of medicinal product indicated in the authorisation, the authorisation for the distribution shall not be valid for the repeated importation of medicinal products. 162. The authorisation for the distribution of parallel imported medicinal products in the Republic of Latvia which has been issued by the State Agency of Medicines until the day of coming into force of this Regulation shall be valid until the expiry date indicated therein. 162.1 Parallel importers which have received the authorisation for the distribution of parallel imported medicinal products in the Republic of Latvia and distribute parallel imported medicinal products in Latvia shall, by 1 October 2010, submit to the State Agency of Medicines a certification that the notification on the intention to commence the distribution of the particular medicinal products indicated in the authorisation in the Republic of Latvia has been provided to the relevant marketing authorisation holder and to owner of the medicinal product trademark (brand). The information referred to in Paragraph 25 of Annex 1 to this Regulation shall be included in the notification. [27 July 2010] 163. The State Agency of Medicines is entitled to re-register the authorisation for the distribution of parallel imported medicinal products on the basis of the submission of the holder (owner) of the authorisation for the distribution of parallel imported medicinal products. The submission shall be submitted to the State Agency of Medicines not later than 60 days prior to the expiration date of the authorisation for the distribution of parallel imported medicinal products. Information indicated in Annex 1 shall be appended to the submission. 164. The authorisation for the distribution of the remaining stock of medicinal products which has been issued by the State Agency of Medicines until the day of coming into force of this Regulation shall be valid until the expiry date indicated therein. 165. The submission for the issuance of the authorisation for the distribution of parallel imported medicinal products in the Republic of Latvia which has been submitted to the State Agency of Medicines until the day of coming into force of this Regulation shall be regarded as valid. 166. The submission for the issuance of the authorisation for the distribution of unregistered medicinal products in the Republic of Latvia which has been submitted to the State Agency of Medicines until the day of coming into force of this Regulation shall be regarded as valid. 167. The fulfilment of the requirements specified in Paragraph 17 of this Regulation shall be ensured until 1 January 2008. 168. The requirement for the submission of the pharmacy request specified in Paragraph 94 of this Regulation to the State Agency of Medicines when an authorisation for the distribution of unregistered medicinal products is being submitted and Sub-paragraph 72.3 of this Regulation shall come into force on 30 December 2007. 169. [27 July 2010] 170. Such pharmacies which distribute medicinal products in retail trade by means of the web and the websites of which are being reconstructed or developed anew shall comply with the requirements specified in Paragraphs 100, 101, 102 and 103 of this Regulation. Other pharmacies for whom a website has been developed shall: 170.1. within one month from the day of coming into force of this Regulation, notify the State Agency of Medicines and the Health Inspectorate of the domain name, electronic mail address, and also amendments thereto; 170.2. correct the content of the website in accordance with the requirements of this Regulation within five months from the day of coming into force of this Regulation. [21 October 2008; 27 July 2010] 171. For the medicinal products which are registered under the centralised registration procedure (the labelling and package leaflet thereof in Latvian has been approved in the European Union) in accordance with Regulation No 726/2004 of the European Parliament and of the Council and which are also registered under the national registration procedure with the State Agency of Medicines, the distribution of the remaining stock of medicinal products registered under the national registration procedure shall be allowed only in the medical treatment institution or social care institution until the remaining stock of medicinal products in such institution has been exhausted. 171.1 Paragraph 67.1 of this Regulation shall come into force on 1 September 2009. [4 August 2009] 171.2 Parallel importers which distribute medicinal products for parallel distribution in the Republic of Latvia for which a certification that the parallel distributor has notified the relevant marketing authorisation holder and the owner of the medicinal product trademark (brand) of the intention to commence the distribution of medicinal products for parallel distribution in the Republic of Latvia has not been submitted to the State Agency of Medicines shall, by 1 October 2010, submit the abovementioned certification to the State Agency of Medicines, indicating particular addressee who has been notified and the date of providing the notification therein. [27 July 2010] 171.3 Sub-paragraph 12.10 of this Regulation shall come into force from 1 January 2011. [27 July 2010] 171.4 The authorisation issued by the State Agency of Medicines: 171.4 1. for the distribution of medicinal products registered in a country of the European Economic Area, but not registered in the Republic of Latvia, which has been issued by the State Agency of Medicines until 1 October 2012 shall be valid until the expiry of the term of validity indicated therein; 171.4 2. for the distribution of unregistered medicinal products for individually granted medicinal products which has been issued by the State Agency of Medicines until 1 October 2012 with an indicated term of validity shall be valid until the expiry of the term of validity indicated therein; 171.4 3. for the distribution of the remaining stock of medicinal products which has been issued by the State Agency of Medicines until 1 October 2012 shall be valid until the expiry of the term of validity indicated therein. [11 September 2012] 171.5 A submission for the receipt of an authorisation for the distribution of medicinal products registered in a country of the European Economic Area, but not registered in the Republic of Latvia, and a submission for the receipt of an authorisation for the distribution of individually granted unregistered medicinal products which has been submitted to the State Agency of Medicines by 1 October 2012 shall be deemed valid. [11 September 2012] 171.6 Sub-paragraph 12.3 1, Paragraphs 12.4 and 113.1 and Sub-paragraph 151.3 of this Regulation shall come into force on 28 October 2013. [8 October 2013] 171.7 Paragraph 66.5 of this Regulation in relation to the issuance of a registration certificate to a person brokering medicinal products, and Sub-paragraph 146.10 of this Regulation in relation to the issuance of a certificate of good distribution practice compliance of medicinal products in the form of a printed document for additional charge shall come into force on 1 July 2014. [8 October 2013] 171.8 The turnover specified in Sub-paragraph 146.13.1 of this Regulation until 31 December 2013 shall be indicated in lats. [8 October 2013] 171.9 Paragraph 53 of this Regulation in relation to the issuance of an authorisation for the distribution of parallel imported medicinal products, Sub-paragraph 79.1 in relation to the issuance of an authorisation for the distribution of the remaining stock of medicinal products, and Paragraph 97 which determines the issuance of an authorisation for the distribution of unregistered medicinal products in the form of a printed document for additional charge, shall come into force on 1 July 2014. [3 December 2013] 171.10 [6 March 2018] 171.11 Sub-paragraphs 12.13.4, 12.16, 12.17, 52.5, 145.5, 146.19, and 153.6 and Paragraphs 63.2 and 67.2 of this Regulation shall be applied from 9 February 2019 in conformity with the transitional measures specified in Articles 48 and 50 of the Delegated Regulation No 2016/161. [15 January 2019] 171.12 The wholesaler of medicinal products and a pharmacy shall, from 9 February 2019, ensure connection with the Repository System of the Medicinal Products of Latvia. [15 January 2019] 171.13 Sub-paragraph 12.18 and Paragraph 149.1 of this Regulation shall come into force on 1 April 2020. The wholesaler of medicinal products which is not distributing the medicinal products included on the list of reimbursable medicinal products shall provide the information referred to in Sub-paragraph 12.18 of this Regulation from 1 July 2020. [17 March 2020] 171.14 Paragraphs 20.1, 20.2, 20.3, 20.4, 20.5, 20.6, 20.7, and 20.8 of this Regulation shall come into force on 1 July 2020. [17 March 2020] 171.15 Amendment to Sub-paragraph 12.5 of this Regulation in respect of the electronic recording of transactions and documents and Paragraphs 12.6 and 12.7 of this Regulation shall come into force on 1 January2021. [17 March 2020; 17 December 2020] 171.16 Amendment to Sub-paragraph 12.14 of this Regulation in respect of the qualification of the responsible person shall come into force on 1 July 2021. [17 March 2020] 171.17 Until 31 December 2021, the general (open) type pharmacy shall remotely process orders of a private individual - medicinal products, including prescription medicinal products, and medicinal products and medical devices reimbursed from the funds of State budget (hereinafter in this Paragraph - the medicinal products) - and delivery thereof to the place of residence of the private individual in conformity with the following requirements: 171.17 1. a general (open) type pharmacy which delivers medicinal products and medical devices to inhabitants at their place of residence shall inform the State Agency of Medicines of the provision of such service, and the State Agency of Medicines shall indicate it on its website. If a person applies to a general (open) type pharmacy which does not provide delivery of the medicinal products, it shall inform the person of the nearest general (open) type pharmacy which provides such service; 171.17 2. a general (open) type pharmacy shall ensure identification of the person - consignee of the medicinal products, remote consultation on the use of the medicinal products, verification of the medicinal products at the pharmacy prior to delivery, and maintenance of the quality of the medicinal products during delivery in conformity with the requirements laid down in the laws and regulations regarding epidemiological safety measures for the containment of the spread of COVID-19 infection; 171.17 3. the delivery of narcotic or psychotropic medicinal products shall be made only by an employee of a general (open) type pharmacy or branch thereof (pharmacist or pharmacist's assistant) or in the case referred to in Section 42 of the Pharmaceutical Law - by a medical practitioner. Restrictions are not laid down for the delivery of other medicinal products to the place of residence of the person in respect of the person who is making it; 171.17 4. medicinal products shall be delivered at home if a person cannot acquire them in person at a pharmacy or delegate another person to receive the medicinal products prescribed in an e-prescription in the unified electronic information system of the health sector; 171.17 5. a general (open) type pharmacy shall accept an order of the medicinal products by phone or using another information society service. During drawing up an order, an employee of the general (open) type pharmacy or branch thereof shall identify a person (consignee of the medicinal products) by his or her given name, surname, address of the place of residence, and telephone number. If a person orders prescription medicinal products to be delivered, the employee of the pharmacy or the branch thereof shall find out also the personal identity number in order to process the e-prescription in the unified electronic information system of the health sector; 171.17 6. the manager of a general (open) type pharmacy shall be responsible for the quality of the delivery service provided. The manager and pharmacist of a general (open) type pharmacy who is organising the issuance and delivery of the medicinal products shall, according to his or her competence, be responsible for the supervision of all stages of the delivery of medicinal products - labelling of medicinal products and attaching a label to the packagings of medicinal products, packaging, delivery of the consignment, and issuance to the person who made the order, and also the provision of consultations on the safe use of medicinal products. [9 June 2020; 17 December 2020; 27 May 2021] 171.18 Until 31 December 2021, without applying the conditions of Paragraphs 34 and 60.2 of this Regulation, the wholesalers of medicinal products shall be permitted to distribute medicinal products in the packaging of the Baltic States (without authorisation for the parallel import or parallel distribution) which is intended for the market of Estonia and Lithuania if they are not available in Latvia, informing thereof the owner of the medicinal product registration certificate. [9 June 2020; 17 December 2020; 27 May 2021] 171.19 Paragraph 11.2 of this Regulation shall come into force on 1 January 2023. [14 December 2021 / Paragraph 11.2 shall be included in the wording of Regulation as of 1 January 2023] 171.20 Until 31 December 2022, the State Agency of Medicines, on the basis of a risk assessment: 171.20 1. may carry out conformity assessment inspections for a medicinal product wholesale facility remotely before granting a special authorisation (licence) or in relation to re-registration of the relevant special authorisation (licence) which is specified in the laws and regulations regarding the licensing of pharmaceutical activity in order to evaluate whether the relevant premises, equipment, devices, staff, and documentation of the medicinal product wholesale facility conform to the requirements laid down in this Regulation and the conditions of special activity at the place where the medicinal products will be stored and from where they will be distributed. If inspections take place remotely, the conditions shall be indicated in the control report (Annex 13.1) in conformity with the course of inspection and the results thereof; 171.20 2. need not carry out conformity assessment inspections at the office premises of medicinal product wholesale facilities if the medicinal products are not stored therein, and if the wholesaler of medicinal products to be inspected has transferred the functions partly to another licensed medicinal product wholesale facility (medicinal product wholesale facility on the basis of a contract), the contractual activities of conformity assessment inspections of wholesalers of medicinal products may be postponed at medicinal product wholesale facilities which are located in the premises of another licensed medicinal product wholesale facility. [14 December 2021] 171.21 After the issuance of the special authorisation (licence) referred to in Paragraph 11 of this Regulation, the State Agency of Medicines may, until 31 December 2022 on the basis of the risk assessment, postpone the inspections of good distribution practice of medicinal products referred to in Sub-paragraphs 146.7 and 146.8 of this Regulation or carry them out remotely. The State Agency of Medicines may postpone the abovementioned inspections if such variations are not intended which expand the scope of the relevant special authorisation (licence) and which must be indicated in a certificate of good distribution practice (for example, new premises, new halls), and also if the competent supervision authority has not implemented activities which affect the validity of the particular certificate of good distribution practice at the premises of the wholesaler of medicinal products located outside the country of the European Economic Area. [14 December 2021] 171.22 If the State Agency of Medicines postpones inspections in the case referred to in Paragraph 171.21 of this Regulation, it shall be regarded that the relevant certificate of good distribution practice is valid until 31 December 2022. [14 December 2021] 171.23 If the State Agency of Medicines remotely carries out the inspections of good distribution practice referred to in Sub-paragraphs 146.7 and 146.8 of this Regulation, the conditions according to the course and results of the inspection shall be indicated in the certificate of good distribution practice issued after inspection. [4 March 2021] 171.24 Until 31 December 2022, the wholesaler of medicinal products may replace the official responsible for conformity with good distribution practice with another person if the responsible official is in quarantine or absent due to the disease caused by COVID-19 or self-isolation, ensuring that the relevant person with whom the responsible official is replaced has appropriate competences, experience, and knowledge in the distribution of medicinal products, he or she has been trained in the issues of good distribution practice, and he or she has appropriate knowledge in the quality management system of the wholesaler of medicinal products. In such case the tasks and duties which are to be performed by the responsible official shall not change but the wholesaler of medicinal products shall notify the State Agency of Medicines of the replacement of the responsible official and append to the notification the curriculum vitae (CV) of the person with whom the responsible official is replaced, and indicate his or her qualification, competences, experience, and knowledge in the distribution of medicinal products, and also training in the issues of good distribution practice. If the State Agency of Medicines recognises that the abovementioned notification of the wholesaler of medicinal products on the particular person cannot be approved, it shall, within 10 working days after receipt of the notification, inform the wholesaler of the necessity to clarify the information or to notify of another person with whom the responsible official is replaced, and indicate the justification for the unfavourable opinion, and also inform the Health Inspectorate thereof. [14 December 2021] 171.25 Paragraph 99.1 and Sub-paragraph 103.1 6 of this Regulation shall come into force on 1 January 2022. [14 December 2021] 172. This Regulation shall come into force on 1 August 2007. Informative Reference to European Union Directives[8 October 2013; 3 December 2013] This Regulation contains legal norms arising from: 1) Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use; 2) Commission Directive 2003/94/EC of 8 October 2003 laying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use (text with EEA relevance); 3) Directive 2004/24/EC of the European Parliament and of the Council of 31 March 2004 amending, as regards traditional herbal medicinal products, Directive 2001/83/EC on the Community code relating to medicinal products for human use; 4) Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use; 5) Directive 2011/62/EU of the European Union and of the Council of 8 June 2011 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use, as regards the prevention of the entry into the legal supply chain of falsified medicinal products; 6) Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010 amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use; 7) Directive 2012/26/EU of the European Parliament and of the Council of 25 October 2012 amending Directive 2001/83/EC as regards pharmacovigilance; 8) Directive 2011/24/EU of the European Parliament and of the Council of 9 March 2011 on the application of patients' rights in cross-border healthcare; 9) Commission Implementing Directive 2012/52/EU of 20 December 2012 laying down measures to facilitate the recognition of medical prescriptions issued in another Member State. Prime Minister A. Kalvītis Acting for the Minister for Health,
Annex 1 [17 March 2020] Submission for Parallel Imported Medicinal Products(Mark with an "x" as appropriate)
Part I
Part II
Part II.A
Differences from the medicinal products registered in Latvia
Part III
Chapter IV (Mark with an "x" as appropriate, indicate the number of pages appended)
Notes. 1. In a column or row, which is not completed, draw a dash. Part III shall be completed if re-packaging is carried out. 2. If information on several medicinal products of the same name with different types of packaging is indicated in one submission, Parts II, II.A, and III shall be prepared separately for each type of packaging. 3. If the submission is submitted for approving variations, only the variations shall be indicated in the relevant box. 4. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents. The time of signing the electronic document is the date and time of attaching the time stamp.
Annex 2 [2 February 2016; 17 March 2020] Authorisation for Distribution of Parallel Imported Medicinal Products in the Republic of LatviaREPUBLIC OF LATVIA STATE AGENCY OF MEDICINES
Distribution of parallel imported medicinal products is permitted:
The parallel imported medicinal products have been re-packaged (mark with an "x" as appropriate):
Place for stamp
Notes. 1. In a column or row, which is not completed, draw a dash. 2. The details of the document "signature" and "place for a seal" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 2.1 Submission for Registration of a Person Brokering Medicinal Products[8 October 2013] To the State Agency of Medicines (mark with an "x" as appropriate)
1. Information on the submitter:
Information on the working hours of such undertaking which brokers medicinal products (the beginning and end of working hours shall be indicated)
2. I request to register variations (mark with an "x" as appropriate):
Appended: 1. Description of the system for the brokering of medicinal products (regarding quality system) on ______ pages. 2. Document by which the merchant or performer of economic activity authorises the contact person referred to in Sub-paragraph 1.6 of this submission to submit a submission and documents to the State Agency of Medicines, on _______ pages. I certify that the information indicated in the submission is complete and true and it conforms to the requirements laid down in Cabinet Regulation No. 416 of 26 June 2007, Distribution and Quality Control of Medicinal Products.
(signature of the person with the right of representation) Notes. 1. If an undertaking has several sites of activity, the information referred to in Sub-paragraph 1.4 of this Annex on other sites and responsible persons shall be indicated on a separate page and appended to the submission. 2. The performer of economic activity who needs not register with the Commercial Register shall indicate the given name, surname and personal identity number in Sub-paragraph 1.1 of this submission, the address of the declared place of residence - in Sub-paragraph 1.2, Sub-paragraph 1.3 need not be completed. 3. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents. 4. Upon submitting a submission for variations, such parts of the form shall be completed to which data on variations apply.
Annex 2.2 [8 October 2013]
Personas, kas veic starpniecības darījumus ar zālēm, reģistrācijas APLIECĪBA Person engaged in brokering of medicinal products REGISTRATION CERTIFICATE 1. Reģistrācijas numurs
__________________________________________ 2. Reģistrētās personas firma
(nosaukums)_____________________________ 3. Reģistrētās personas juridiskā adrese
_____________________________ 4. Faktiskās darbības norises vietas adrese
________________________________________ 5. Reģistrācijas juridiskais pamatojums
________________________________ Šī reģistrācijas apliecība ir derīga tikai pilnā apjomā,
iekļaujot visas lapas. Šīs reģistrācijas apliecības 2.punktā minētā persona par
jebkurām izmaiņām ziņo nekavējoties.
Piezīme. Dokumenta rekvizītu "paraksts" neaizpilda, ja
elektroniskais dokuments ir sagatavots atbilstoši normatīvajiem
aktiem par elektronisko dokumentu noformēšanu.
Annex 3 Distribution Authorisation for Selling Remaining Stock of Medicinal Products[2 February 2016]
Annex 4 [11 September 2012; 8 October 2013; 17 March 2020] REPUBLIC OF LATVIA STATE AGENCY OF MEDICINES
Authorisation for Distribution of Medicinal Products Registered in a Country of European Economic Area but Unregistered in the Republic of LatviaRīga
on the issuance of the authorisation for medicinal products registered in a country of the European Economic Area, but unregistered in the Republic of Latvia, for the purpose of protection of public health it is permitted to distribute the following medicinal products in the Republic of Latvia:
Bringing in of medicinal products from countries of European Economic Area is permitted.
Place for a seal Notes. 1. In column 3 of the Table the marketing authorisation holder or the submitter of the submission for registration of the medicinal product shall be indicated in accordance with the laws and regulations regarding the procedures for registering medicinal products. 2. In column 6 of the Table the anatomical therapeutic chemical classification code (ATC code) of the medicinal product shall be indicated. 3. In column 7 of the Table the identification number of medicinal products assigned by the State Agency of Medicines for each size of the packaging of the form of medicinal products shall be indicated. 4. In column 8 of the Table the classification group which has been granted in accordance with the laws and regulations regarding the procedures for the classification of medicinal products, and the designation thereof shall be indicated. The designation shall not be indicated for medicinal products not subject to medical prescription. 5. In a column or row, which is not completed, a dash shall be drawn. 6. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 5 [11 September 2012; 8 October 2013; 2 February 2016] Submission for Receipt of an Authorisation for Distribution of Medicinal Products Registered in a Country of European Economic Area but Unregistered in the Republic of Latvia(Mark with an "x" as appropriate) Submission for granting an authorisation Submission for variations in documentation We request the State Agency of Medicines to issue an authorisation for the distribution of the unregistered medicinal products indicated in Part I of the submission in the Republic of Latvia. We wish to receive the authorisation in the form of a printed document (mark with an "x" as appropriate)
Part I
Part II (mark with an "x" as appropriate, indicate the number of pages appended)
certify that the information provided by me is true.
Notes. 1. In a column or row, which is not completed, a dash shall be drawn. 2. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 6 [11 September 2012; 8 October 2013; 2 February 2016; 17 March 2020] REPUBLIC OF LATVIA STATE AGENCY OF MEDICINES
Authorisation for Distribution of Unregistered Medicinal Products for Individually Granted Medicinal Products
On the basis of the decision No.__ of the State Agency of Medicines on the issuance of the authorisation for the distribution of unregistered medicinal products in the Republic of Latvia, the distribution of medicinal products in the Republic of Latvia is permitted to
Place for a seal If the quantity of the packaging units of medicinal products is indicated in the authorisation, a new authorisation for the distribution of unregistered medicinal products is necessary for repeated distribution of the medicinal products according to the quantity of packaging units of medicinal products indicated in the authorisation. Notes. 1. In a column or row, which is not completed, a dash shall be drawn. 2. In the Table, the section "Bringing in of medicinal products is permitted from:" shall be filled in according to the information indicated in the submission for the receipt of an authorisation for the distribution of individually granted unregistered medicinal products. 3. In column 4 of the Table the person to whom the authorisation holder is entitled to distribute the medicinal products indicated in the authorisation (shall not apply to the authorisation referred to in Notes 4 and 5) shall be indicated, by including an indication: 3.1. "to a pharmacy"; 3.2. "to the medical treatment institution", and indicate its name and registration number in the Register of Medical Treatment Institutions; 3.3. "to the social care institution", and indicate its name, number of the taxpayer certificate of the State Revenue Service in the Register of Taxpayers of the State Revenue Service; 3.4. "to the practising veterinarian", and indicate his or her given name, surname and registration number in the Register of Enterprises Subject to Monitoring by the Food and Veterinary Service; 3.5. "to the institution engaged in veterinary medical care", and indicate its name and registration number in the Register of Enterprises Subject to Monitoring by the Food and Veterinary Service. 4. For the medicinal products unregistered in the authorisation which conform to the list of medicinal products to be used or which are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient treatment, or on which there is a request of a medical treatment institution, social care institution, practising veterinarian, or veterinary medical care institution for the acquisition of medicinal products to which an opinion of the professional association of doctors or professional panel of veterinarians has been appended respectively (shall not apply to the authorisation referred to in Note 3): 4.1. column 3 of the Table shall not be completed; 4.2. in column 4 of the Table the relevant indication shall be included: 4.2.1. to be distributed only to hospitals and closed type pharmacies or pharmacies of medical treatment institutions and general or open type pharmacies (applies to unregistered medicinal products which are included in the list of usable medicinal products); 4.2.2. to be distributed only to general type pharmacies (applies to unregistered medicinal products which are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient treatment); 4.2.3. to be distributed to pharmacies, medical treatment institutions, and social care institutions, practising veterinarians or veterinary medical care institutions (applies to unregistered medicinal products on which there is a request of a medical treatment institution or social care institution for the acquisition of medicinal products to which an opinion of the professional association of doctors has been appended or a request of a veterinary medical care institution or practising veterinarian for the acquisition of medicinal products to which an opinion of the professional panel of veterinarians has been appended). 5. For medicinal products for compassionate use: 5.1. in column 4 "Medicinal product may be distributed to" of the Table the following shall be included: 5.1.1. the indication "Medicinal products available for compassionate use"; 5.1.2. the name of the medical treatment institution to which medicinal products are delivered and their registration number in the Register of Medicinal Treatment Institutions of the Health Inspectorate; 5.2. in column 5 of the Table the following shall be indicated: 5.2.1. the name, address of the manufacturer of the medicinal product, contact persons (given name, surname, position, telephone, fax, and electronic mail address (if any)); 5.2.2. the name of the country of manufacture; 5.2.3. the name, address of the authorisation requester, and contact persons (given name, surname, position, telephone, fax, electronic mail address (if any)). 6. In the introductory part of the authorisation, after the entry on the name and type of the legal person, the number of the special authorisation (licence) shall be indicated for the medicinal product wholesale facility and medicinal product manufacturer instead of the registration number. 7. In column 1 of the Table the identification number of the medicinal product granted by the State Agency of Medicines shall be indicated. 8. The indication "The authorisation is valid until" shall be included in the authorisation, if the authorisation is issued for: 8.1. medicinal products conforming to the list of usable medicinal products; 8.2. medicinal products which are distributed within the scope of the system for the reimbursement of expenses for the acquisition of medicinal products intended for out-patient treatment; 8.3. medicinal products for compassionate use if the term of validity of the authorisation is determined; 9. The indication "After distribution of the quantity of the medicinal products indicated in the authorisation, a new authorisation for distribution of unregistered medicinal products is necessary for repeated distribution (importation) of the medicinal products" shall be included in the authorisation, if the quantity of the packaging units is indicated in the authorisation. In order to ensure availability of medicinal products, the number of packagings indicated in the authorisation may be greater than that indicated in the request of pharmacies for the acquisition of medicinal products if the medicinal product wholesale facility has indicated it in the submission for obtaining an authorisation. 10. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 7 [27 July 2010; 11 September 2012; 8 October 2013; 2 February 2016; 17 March 2020] Submission for Obtaining an Authorisation for Distribution of Individually Granted Unregistered Medicinal ProductsWe request the State Agency of Medicines to issue an authorisation for the distribution of individually granted unregistered medicinal products in the Republic of Latvia for the medicinal products indicated in Part I. We wish to receive the authorisation in the form of a printed document (mark with an "x" as appropriate)
Part I
Part II Mark as appropriate with an "x" and indicate the number of pages appended
certify that the information provided is true.
Date of the receipt of the submission at the State Agency of Medicines ______________________ Notes. 1. The licence number shall not be indicated in Sub-paragraph 1.1 of Part I, if the authorisation is requested for medicinal products for compassionate use. In this case, the registration number of the medical treatment institution in the Register of Medical Treatment Institutions shall be indicated in Sub-paragraph 1.1 of Part I. 2. In a column or row, which is not completed, a dash shall be drawn. 3. If the form is sent without using electronic data media, the applicant shall sign each page appended to the form. 4. [11 September 2012] 5. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 8 [11 September 2012] Medicinal Products Subjected to Quality ControlThe following medicinal products are subject to quality control:
Note. The control referred to in Sub-paragraphs 1.1, 2.1 and 3.1, Paragraphs 4 and 5 and Sub-paragraph 6.1 of this Annex shall also be applied to the wholesaler of medicinal products which imports medicinal products (a person who has the special authorisation (licence) referred to in Sub-paragraph 11.2 of this Regulation for the manufacturing or importing medicinal products). Acting for the Minister for Health,
Annex 8.1 [2 February 2016] Notification of a Marketing Authorisation Holder on the Activity Executed Thereby to Suspend Distribution of Medicinal Products, to Request Recall of Medicinal Products from the Market, to Request Cancellation of Registration or not to Apply for the Re-registration of Medicinal Products, and on the Justification for the Relevant ActivityNotification of "withdrawn products" by marketing authorization holders
Apliecinu, ka kopā ar šo pavadvēstuli tiek iesniegta izplatīšanas lapa MS ExcelM formātā (veidne EMA/445787/2013), nosaukta <Zāļu reģistrācijas īpašnieka reģistrācijas faila nosaukums>, kas satur visu nepieciešamo informāciju par attiecīgajām zālēm. I herewith confirm that together with this cover letter an Excel spread sheet (template EMA/445787/2013) entitled <MAH file name> is submitted containing all the required information related to the medicinal product(s) concerned. Ar cieņu Yours sincerely
Notes. 1. Paziņojuma pavadvēstuli noformē uz reģistrācijas apliecības īpašnieka veidlapas. The cover letter of the notification has to be written on a headed paper. 2. Dokumenta rekvizītu "paraksts" neaizpilda, ja elektroniskais dokuments ir sagatavots atbilstoši normatīvajiem aktiem par elektronisko dokumentu noformēšanu. The document part "Signature" shouldn`t be filled in if the electronic document is prepared in accordance with the normative acts on creation of electronic documents. 3. Eiropas zāļu aģentūrai paziņojumu nosūta uz elektroniskā pasta adresi: withdrawnproducts@ema.europa.eu. European Medicines Agency notice shall be sent to the e-mail address: withdrawnproducts@ema.europa.eu
Annex 9 Quality Defects of Medicinal Products1. Class I defects are potentially life threatening or may cause a serious risk to health, for example: 1.1. wrong medicinal product (label and contents are different products); 1.2. correct medicinal product but wrong strength, with serious medical consequences; 1.3. microbiological contamination of a sterile injectable or ophthalmic medicinal product; 1.4. chemical contamination with serious medical consequences; 1.5. mix-up of some types of products within more than one container; 1.6. wrong active ingredient in a multi-component medicinal product, with serious medical consequences. 2. Class II defects may cause illness or mistreatment, but are not classified as Class I defects, for example: 2.1. poor labelling, for example, wrong or missing text or figures; 2.2. missing or incorrect information (package leaflet); 2.3. microbiological contamination with medical consequences of a non-injectable liquid, sterile medicinal product which is used in ophthalmology; 2.4. chemical or physical contamination (significant impurities, cross-contamination, particulates); 2.5. mixing in of some products in containers (rogues); 2.6. non-compliance with specifications (for example, assay, stability, fill, weight); 2.7. insecure closure with serious medical consequences (for example, cytotoxics, child-resistant container, potent medicinal products). 3. Class III defects may not pose a significant hazard to health, but withdrawal may be initiated for other reasons, for example: 3.1. faulty packaging (for example, wrong or missing batch number or expiry date); 3.2. faulty closure; 3.3. contamination of outer packaging (for example, microbiological spoilage, dirt or detritus (dead organic waste) material, particulate matter. Acting for the Minister for Health,
Annex 10 [21 October 2008; 27 July 2010; 8 October 2013] Notification on Recall of Medicinal ProductsI. Notification provider:
II. Information on recalled medicinal products
Note. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents. Acting for the Minister for Health,
Annex 11 Notification of Continuous Operation Telephone Number (or Other Means of Communication) Available for Communication Twenty-four Hours a Day and Contact Persons Responsible for the Recall of Medicinal Products[2 February 2016]
Annex 12 [21 October 2008; 27 July 2010; 8 October 2013]
Notes. 1. In a column or row, which is not completed, draw a dash. 2. If the authorisation is drawn up on several pages, the responsible official shall sign each page. 3. If an order is issued that after the initial rapid alert the batch needs a following rapid alert, it shall be indicated in Paragraph 18 "Rapid Alert following original rapid alert# ref.no#". To be filled in by the Health Inspectorate. 4. The rapid alert notification shall have a reference number, which is formed from the country code (country in which the fact is establish the first time), the code of the competent authority, and, where necessary, classification and correspondence number (for example, LV//II/04/07 - indicates Class II rapid alert initiative from Latvia, the fourth alert in succession which has been initiated by Latvia, the seventh rapid alert correspondence (in conformity with the requirements of Chapter XII of Cabinet Regulation No.416 of 26 June 2007, Procedures for the Distribution and Quality Control of Medicinal Products). To be filled in by the Health Inspectorate. 5. If the notification is provided for counterfeit medicinal products, the title shall indicate "Rapid Alert Notification on Counterfeit Medicinal Product". 6. The owner of the special authorisation (licence) for medicinal product manufacturing and the qualified person who has released the batch in the name of the owner of the special authorisation (licence) shall be indicated in Paragraph 15, if they are not one and the same person. 7. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents. Acting for the Minister for Health,
Annex 13 [27 July 2010; 8 October 2013]
Note. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents. Acting for the Minister for Health,
Annex 13.1 [2 February 2016] Report On Inspection of Good Distribution Practice of Medicinal ProductsRiga
1. Introductory part 1.1. Short description of the undertaking and entrepreneurial activities The fields of activity of the undertaking and the types of products distributed shall be indicated. 1.2. Previous inspection Date, given name, surname of the inspector who participated in the previous inspection. 1.3. Variations since the previous inspection The most significant variations in the staff, premises, installations and equipment shall be described. If possible, references shall be indicated or information shall be included from documents of the undertaking in which these or future changes are described. The principal text of the report must be written in the past form because the report applies to that observed during inspection. Other verb tense may be used in the description of future plans. 2. Short layout of the inspection activities carried out 2.1. Field of inspection (type and reason) Short description of inspection (for example, conformity of distribution activities with good distribution practice). The reason for inspection shall be indicated (for example, repeat (routine) inspection, submission for the receipt of a licence, justified reason for inspection). 2.2. Purpose of inspection 2.3. Inspected area and activities Each activity inspected shall be indicated. 2.4. Non-inspected areas and activities If necessary, the area or activities which were not inspected, shall be indicated. 2.5. The staff met during the inspection 2.6. The responsible person The given name, surname, name of the position of those representative of the managing staff shall be indicated with whom meetings took place during the inspection. 3. Observations of inspectors in relation to the inspection and deficiencies detected References to the titles of the relevant chapters of the good distribution practice guidelines shall be indicated. A short description of the relevant activities must be indicated within the scope of the section. The most significant procedures or aspects, as well as future plans which might affect the performance of the next inspection, may be indicated. This section may be linked to the section regarding the deficiencies detected in order to explain their classification. Report on the measures detected in the previous inspection and taken for the elimination of deficiencies Quality management Staff Premises and installations Documentation Activities Complaints, return of medicinal products, suspicions of counterfeit medicinal products, and recall of medicinal products Activities transferred to providers of outsourced services Self-inspection Transportation Other specific issues identified For example, significant variations in the future notified by the undertaking. 4. Various information Samples taken Annexes appended. Information on the administrative punishments imposed for violating the specified procedures in pharmaceutical activity or violating the laws and regulations regarding circulation of narcotic and psychotropic substances and medicinal products after the previous inspection, the officials punished, and information on the payment of a fine shall also be indicated in Annex. List of annexes appended List of deficiencies (critical, significant and other) All deficiencies and a corresponding reference to the requirements of the laws and regulations governing the field of distribution of medicinal products shall be indicated. All deficiencies detected shall be indicated also in case, if they were eliminated at one. The undertaking shall be requested to inform the responsible authorities of the deadlines for and progress of measures for the elimination of deficiencies. Each deficiency, if possible, shall be indicated in the form of a negation. Deficiencies must be indicated in a short and concise manner, for example, without using the phrase "approach to monitoring of the temperature did not conform to the requirements of good distribution practice", but: "approach to monitoring of the temperature did not conform to the requirements of good manufacturing practice because: • the devices for measuring temperature were not calibrated; • records of temperature data were not regularly reviewed". In defining the deficiencies, such words as "non-conforming", "insufficient", "inappropriate" must be used. 5. Comments of the inspector May be used, for example, to indicate actual information or orally expressed promises received during the inspection, or to indicate whether the answers provided by the undertaking are acceptable. 6. Recommendations for further activities Recommendations for the undertaking or institution shall be provided, if any. 7. Summary and conclusions The inspector shall indicate whether the distributor is operating in the field inspected according to the guidelines for good distribution practice of medicinal products published by the European Commission. If necessary, he or she shall indicate whether appropriate corrective and preventive actions have been taken, as well as any other information which would attract the attention of the institution which requested the performance of inspection. The report shall be signed and date by all inspectors/experts who participated in the inspection. Given name, surname Signature Institution Date Sending of the report Notes. 1. Definitions of significant deficiencies: 1.1. critical deficiencies - any deviations from the requirements of the guidelines for good distribution practice due to which the product may become or has become harmful to human or public health. Critical deficiency is also an aggregate of significant deficiencies which points towards a serious inability of the system to ensure appropriate operation. For example, the following may be a critical deficiency: 1.1.1. the acquisition of medicinal products from an undertaking or delivery to an undertaking to which a special authorisation (licence) for the wholesale trade of medicinal products has not been issued; 1.1.2. the storage of medicinal products at room temperature if storage at a low temperature has been specified for them; 1.1.3. the rejected or recalled medicinal products are in the stock to be sold; 1.2. significant deficiencies - deficiencies which are not critical deficiencies and: 1.2.1. which point towards large deviations from the guidelines of good distribution practice; 1.2.2. due to which the product does not conform to the documentation, particularly to the storage and transportation circumstances; 1.2.3. which point towards large deviations from the conditions of the special authorisation (licence) for wholesale trade of medicinal products (within the scope of the European Union and the European Economic Area); 1.2.4. combination of various other deficiencies from which each individual deficiency is not significant, but together they may cause significant deficiency, therefore, they must be explained and they should be notified as a significant deficiency; 1.3. other deficiencies - deficiencies which may not be classified as critical or significant, but which point towards deviations from the guidelines of good distribution practice. Deficiencies may be classified as "other deficiencies" if they are evaluated as non-significant or there is not sufficient information to classify them as significant or critical. 2. The detail of the document "signature" shall not be completed if the electronic document has been prepared in accordance with the laws and regulations regarding drawing up of electronic documents.
Annex 14 [2 February 2016]
ZĀĻU IZPLATĪTĀJA LABAS IZPLATĪŠANAS PRAKSES ATBILSTĪBAS SERTIFIKĀTS ATTIECĪBĀ UZ CILVĒKIEM PAREDZĒTAJĀM ZĀLĒM CERTIFICATE OF GDP COMPLIANCE OF A DISTRIBUTOR OF MEDICINAL PRODUCTS FOR HUMAN USE
iegūtā informācija ļauj uzskatīt, ka tas atbilst labas izplatīšanas prakses principiem, kas minēti Direktīvas 2001/83/EK 84. pantā. From the knowledge gained during inspection of this distributor, the latest of which was conducted on …../...…/...… [date], it is considered that it complies with the principles of good distribution practice requirements laid down in Article 84 of Directive 2001/83/EC. Šis sertifikāts atspoguļo izplatīšanas vietas statusu minētās oficiālās pārbaudes laikā, un tas nevar atspoguļot atbilstības statusu, ja ir pagājuši vairāk nekā pieci gadi kopš oficiālās pārbaudes, kad tika izsniegts šis sertifikāts. Derīguma termiņš var tikt saīsināts, izmantojot riska vadības regulējošos principus un izdarot ierakstu lauciņā, kas atvēlēts ierobežojumu vai paskaidrojumu atzīmēšanai. This certificate reflects the status of the premises at the time of the inspection noted above and should not be relied upon to reflect the compliance status if more than five years have elapsed since the date of official inspection, after which time the issuing authority should be consulted. This period of validity may be reduced, using regulatory risk management principles by an entry in the "Restrictions" or "Clarifying Remarks" field. Šis sertifikāts ir derīgs tikai pilnā apjomā, uzrādot visas lapas. This certificate is valid only when presented with all pages. Sertifikāta autentiskumu var pārbaudīt Savienības datubāzē. Ja sertifikāta nav datubāzē, lūdzu, sazinieties ar Zāļu valsts aģentūru. The authenticity of this certificate may be verified in the Union's database. If it does not appear, please contact the issuing authority. Ierobežojumi vai paskaidrojumi saistībā ar šā sertifikāta jomu: Any restrictions or clarifying remarks related to the scope of this certificate:
Informācija par licenci pieejama Kopienas datubāzē. Details of the authorisation can be found in the Union database. Notes. 1. Paraksts, datums un kontaktinformācija ir uz katras sertifikāta lappuses. The signature, date and contact details should appear on each page of the certificate. 2. Dokumenta rekvizītu "paraksts" neaizpilda, ja elektroniskais dokuments ir sagatavots atbilstoši normatīvajiem aktiem par elektronisko dokumentu noformēšanu. Document property "Signature" is not filled in if the document is prepared in accordance with the laws of electronic documents. 3. Šajā sertifikātā minētā Savienības datubāze, kurā pārbauda sertifikāta autentiskumu, ir Eiropas Savienības datubāze par ražošanas un importēšanas licencēm un labas ražošanas prakses sertifikātiem (Eudra GMDP datubāze). The Union database mentioned in this certificate is the European Union database on the production and import licenses and certificates of good manufacturing practice (Eudra GMDP database). Translation © 2023 Valsts valodas centrs (State Language Centre) |
Document information
Title: Zāļu izplatīšanas un kvalitātes kontroles kārtība
Status:
In force
Language:   Related documents
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Latvijas Vestnesis, the official publisher ensures legislative acts systematization function on this site. All Likumi.lv content is intended for information purposes. |
About Likumi.lv News archive For feedback Apmācības Contacts |
Mobile version Terms of service Privacy policy Cookies Accessibility |
|
 yes
yes