Text consolidated by Valsts valodas centrs (State
Language Centre) with amending regulations of:
10 June 2008 [shall come
into force on 14 June 2008];
29 September 2009 [shall come into force on 3 October
2009];
14 September 2010 [shall come into force on 18 September
2010];
13 August 2013 [shall come into force on 16 August
2013];
2 February 2016 [shall come into force on 12 February
2016];
25 September 2018 [shall come into force on 28 September
2018];
15 January 2019 [shall come into force on 18 January
2019];
14 July 2022 [shall come into force on 19 July 2022].
If a whole or part of a paragraph has been amended,
the date of the amending regulation appears in square
brackets at the end of the paragraph. If a whole
paragraph or sub-paragraph has been deleted, the date of
the deletion appears in square brackets beside the
deleted paragraph or sub-paragraph.
|
|
Republic of Latvia
Cabinet
Regulation No. 436
Adopted 26 June 2007
|
Procedures for the Importation and
Exportation of Medicinal Products
Issued pursuant to
Section 5, Clause 3 of the Pharmaceutical Law
and Section 28 of the Law on the Legal Trade
of Narcotic and Psychotropic Substances
and Medicinal Products, and also Precursors
[14 July 2022]
I. General Provisions
1. The Regulation prescribes:
1.1. the procedures for the importation and exportation of
medicinal products;
1.2. the customs control points (hereinafter - the border
crossing points through which the import and export of the
substances and medicinal products included in Schedules II and
III of Narcotic Substances, Psychotropic Substances and
Precursors to be Controlled in Latvia;
1.3. the supervision of the importation and exportation of
medicinal products.
[14 July 2022]
2. The Regulation applies to the importation into the customs
territory of the European Union of these medicinal products and
auxiliary medicinal products (hereinafter - the medicinal
products) (imports of medicinal products) and their exportation
from the customs territory of the European Union (exports of
medicinal products):
2.1. the import of such medicinal products which have been
registered in the Register of Medicinal Products of the Republic
of Latvia or under the centralised authorisation procedure in
accordance with Regulation (EC) No 726/2004 of the European
Parliament and of the Council of 31 March 2004 laying down
Community procedures for the authorisation and supervision of
medicinal products for human use and establishing a European
Medicines Agency (hereinafter - Regulation No 726/2004 of the
European Parliament and of the Council);
2.2. the import of such medicinal products that have not been
registered in the Register of Medicinal Products of the Republic
of Latvia or under the centralised authorisation procedure in
accordance with Regulation (EC) No 726/2004 of the European
Parliament and of the Council, but have been registered in third
countries (hereinafter - the unregistered medicinal products from
third countries);
2.3. the import of medicinal products by budget institutions
or public benefit organisations in accordance with Council
Regulation (EC) No 1186/2009 of 16 November 2009 setting up a
Community system of reliefs from customs duty (does not apply to
auxiliary medicinal products);
2.4. the import of the samples of medicinal products,
including substances used as reference substances for the testing
of medicinal products (hereinafter - the samples of reference
standards);
2.5. the export of medicinal products and sample medicinal
products, including samples of reference standards;
2.6. the import and export of investigational medicinal
products.
[14 July 2022]
2.1 In free ports, special economic zones and
customs warehouses, and also in other storage places for
medicinal products referred to in Paragraph 2 of the Regulation,
the entry (import) and exit (export) of medicinal products shall
be subject to supervision in accordance with the Regulation.
[14 July 2022]
2.2 The importation of investigational medicinal
products (hereinafter - the investigational medicinal products)
specified in Article 2(2)(5) of Regulation (EC) No 536/2014 of
the European Parliament and of the Council of 16 April 2014 on
clinical trials on medicinal products for human use and repealing
Directive 2001/20/EC (hereinafter - Regulation No 536/2014 of the
European Parliament and of the Council), and the import of
auxiliary medicinal products specified in Article 2(2)(8) of
Regulation No 536/2014 of the European Parliament and of the
Council shall be carried out in accordance with Chapter IX of
Regulation No 536/2014 of the European Parliament and of the
Council and this Regulation.
[14 July 2022]
3. This Regulation shall not apply to:
3.1. the importation of medicinal products from countries of
the European Economic Area or the exportation of medicinal
products to countries of the European Economic Area;
3.2. the importation and exportation of medicinal products for
personal use by a natural person, for example, to medicinal
products in the luggage of a traveller;
3.3. medicinal products which a natural person receives or
sends as postal items;
3.4. veterinary medicinal products.
[14 September 2010; 14 July 2022]
4. The import and export of the medicinal products referred to
in Paragraph 2 of this Regulation (including medicinal products
containing the substances (narcotic medicinal products) included
in Schedule II of Narcotic Substances, Psychotropic Substances
and Precursors to be Controlled in Latvia and the substances
(psychotropic medicinal products) included in Schedule III of
Narcotic Substances, Psychotropic Substances and Precursors to be
Controlled in Latvia) are permitted through border crossing
points in which the Food and Veterinary Service performs food
safety and non-food product safety, quality and classification
checks in relation to compliance with the temperature regime
which are specified in the laws and regulations regarding border
crossing points and the inspections to be carried out
therein.
[14 July 2022]
5. The importation and exportation of medicinal products shall
be controlled by the customs authorities in accordance with the
Customs Law and the relevant laws and regulations that govern the
procedures for customs clearance and customs control.
5.1 The Health Inspectorate shall carry out market
surveillance as regards the medicinal products for human use in
accordance with the Pharmaceutical Law, the laws and regulations
regarding the distribution and quality control of medicinal
products and this Regulation.
[14 July 2022]
6. The Food and Veterinary Service shall carry out documentary
checks and also, on the basis of risk analysis, identity and
physical checks in accordance with:
6.1. this Regulation;
6.2. Regulation No 2016/793 of the European Parliament and of
the Council of 11 May 2016 to avoid trade diversion into the
European Union of certain key medicines (codification)
(hereinafter - Regulation No 2016/793 of the European Parliament
and of the Council);
6.3. Article 14 of Regulation (EC) No 816/2006 of the European
Parliament and of the Council of 17 May 2006 on compulsory
licensing of patents relating to the manufacture of
pharmaceutical products for export to countries with public
health problems (hereinafter - Regulation No 816/2006 of the
European Parliament and of the Council).
[14 July 2022]
7. [1 January 2023 / See Paragraph 56.3]
7.1 The Health Inspectorate shall, prior to the
issuing or re-registration of a special authorisation (licence),
inspect the conformity of such customs warehouses where medicinal
products are intended to be stored for more than 72 hours,
premises, equipment, facilities and personnel with the
requirements specified in the laws and regulations regarding the
distribution of medicinal products. After inspection, the Health
Inspectorate shall prepare and issue an inspection deed to the
holder of the customs warehouse permit, and also submit it to the
State Agency of Medicines.
[14 July 2022]
7.2 If the medicinal products are stored in a
customs warehouse for up to 72 hours or at a place of temporary
storage, the holder of the authorisation for the relevant customs
warehouse and the holder of the authorisation for the temporary
storage facility has the obligation to notify the Health
Inspectorate thereof prior to the commencement of such service.
The abovementioned notice shall not apply to the customs
warehouses to which the Health Inspectorate has issued an opinion
on the conformity of the warehouse with the requirements for the
storage of medicinal products and to the customs warehouses
specified in the special authorisation (licence) of the medicinal
product wholesaler.
[14 July 2022 / Paragraph shall come into force on 1
January 2023. See Paragraph 56.4]
8. The owner or the possessor (hereinafter - the possessor of
medicinal products) of a freight of medicinal products shall:
8.1. submit an instruction to the holder of the authorisation
for a customs warehouse and the holder of the authorisation for a
temporary storage facility, specifying the requirements for the
storage of medicinal products in accordance with the storage
conditions specified by the medicinal product manufacturer
(instructions for use of medicinal products, descriptions of
medicinal products). The holder of the authorisation for a
customs warehouse and holder of the authorisation for a temporary
storage facility shall ensure conditions for the storage of
medicinal products in the customs warehouse and temporary storage
facility accordingly in compliance with the instructions of the
possessor of medicinal products and requirements for the storage
of medicinal products in accordance with the guidelines for good
distribution practice of medicinal products published by the
European Commission (available in the official language on the
website of the State Agency of Medicines);
8.2. cover the expenditures that are related to the ensuring
of the conditions for the storage of medicinal products in the
customs warehouse;
8.3. when importing medicinal products, present them at the
places for checks referred to in Paragraph 4 of this Regulation
for the check of the Food and Veterinary Service and ensure free
access for the officials of the Health Inspectorate to the place
where medicinal products are stored in customs warehouses and
temporary storage facilities.
[10 June 2008; 29 September 2009; 14 September 2010; 2
February 2016; 14 July 2022]
9. An accompanying document issued by the relevant foreign
country shall be attached by the possessor of medicinal products
to the freight of medicinal products that is imported, and the
following information shall be indicated therein:
9.1. the date of the delivery of medicinal products, name of
the medicinal products, pharmaceutical form, strength or
concentration of the medicinal products and the manufacturing
batch number and amount of each medicinal product supplied, the
firm name and address of the supplier of the medicinal products
(consignor), the firm name of the medicinal product manufacturer,
the name of the country of manufacture of the medicinal products
and the firm name and address of the consignee of the medicinal
products;
9.2. the price for which the medicinal products have been sold
to the consignee of the medicinal products.
[13 August 2013]
10. If the possessor of medicinal products uses, on the basis
of a contract, transport services provided by another person
(hereinafter - the commercial carrier) for the import of freight,
then the commercial carrier shall, in addition to the
requirements laid down in Paragraph 9 of this Regulation, present
to the customs authority the contract concluded between the
possessor of medicinal products and the commercial carrier for
the provision of transport services or the authorisation of the
possessor of medicinal products for the performance of the
relevant activity.
II. Import of Medicinal
Products
11. Medicinal products may be imported by a person who,
according to the laws and regulations regarding the procedures
for the licensing of pharmaceutical activity, has a special
authorisation (licence) issued by the State Agency of Medicines
for the manufacture or importation of medicinal products with the
authorised activity - import of medicinal products (this shall
not apply to unregistered medicinal products, samples of
medicinal product, and transit of medicinal products - freight of
medicinal products that enters from third countries into the
places referred to in Paragraph 2.1 of this Regulation
and is exported to third countries). Investigational medicinal
products may be imported by a person in the special authorisation
(licence) for the manufacture/importation of medicinal products
of whom it is indicated that the import of investigational
medicinal products is permitted.
[14 July 2022]
11.1 The special authorisation (licence) for the
manufacture and importation of medicinal products issued by the
State Agency of Medicines shall not be necessary for a freight of
medicinal products that is imported from third countries on the
basis of a licence for the manufacture and importation of
medicinal products issued by the competent authority of another
European Union Member State and that is transported in transit
(including placed in a customs warehouse) through the territory
of Latvia.
[14 July 2022]
12. A person who is involved in the activities for the
performance of which the special authorisation (licence) for the
manufacture/importing of medicinal products (hereinafter - the
importer of medicinal products) is necessary shall ensure the
fulfilment of the following requirements:
12.1. the imported medicinal products, including the
investigational medicinal products, have been manufactured in
compliance with the requirements that are equivalent to or higher
than the principles of and guidelines for good manufacturing
practice specified in the Pharmaceutical Law and laws and
regulations regarding the procedures for the manufacture and
control of medicinal products. Investigational medicinal products
are subject to the principles of and guidelines for good
manufacturing practice referred to in Article 63 of Regulation No
536/2014 of the European Parliament and of the Council under
Chapter II of Commission Delegated Regulation (EU) 2017/1569 of
23 May 2017 supplementing Regulation (EU) No 536/2014 of the
European Parliament and of the Council by specifying principles
of and guidelines for good manufacturing practice for
investigational medicinal products for human use and arrangements
for inspections (hereinafter - Commission Delegated Regulation No
2017/1569);
12.2. the medicinal product manufacturer has a relevant
authorisation for the manufacture of medicinal products in the
relevant country;
12.3. at least one responsible official with corresponding
education and professional experience (hereinafter - the
qualified person) is permanently and continuously at the disposal
thereof. The State Agency of Medicines shall be immediately, but
not later than within five days, must be notified in writing of
the change of the qualified person;
12.4. a personnel it at the disposal thereof which meets the
requirements laid down in the laws and regulations regarding the
manufacture and control of medicinal products;
12.5. a possibility to visit the premises of the importer of
medicinal products at any time is ensured to officials of the
State Agency of Medicines and the Health Inspectorate;
12.6. the qualified person is provided with the possibility to
fulfil the requirements referred to in Paragraphs 14, 15 and 16
of this Regulation (in relation to investigational medicinal
products - the requirements referred to in Paragraphs 21 and 22
of this Regulation), for example, by placing at the disposal
thereof the necessary facilities;
12.7. the principles of and guidelines for good manufacturing
practice that have been specified in the laws and regulations
regarding the manufacture and control of medicinal products is
complied with in the quality control and batch release of the
imported medicinal products;
12.8. in the distribution of medicinal products, principles of
good distribution practice of medicinal products laid down in the
guidelines for good distribution practice of medicinal products
published by the European Commission (available in the official
language on the website of the State Agency of Medicines) are
followed. The requirements laid down in the laws and regulations
for the conducting the clinical trials of medicinal products must
be complied with in relation to investigational medicinal
products.
[2 February 2016; 25 September 2018; 14 July 2022]
13. The education and professional experience of the qualified
person shall conform to the qualification and professional
experience criteria that have been determined in the laws and
regulations regarding the manufacture and control of medicinal
products.
14. The qualified person shall, without prejudice to his or
her relationship with the importer of medicinal products, be
responsible for ensuring that a full qualitative analysis and
quantitative analysis of all active substances is carried out for
each batch of imported medicinal products (also if the medicinal
products have been manufactured within the European Community
(Member States of the European Union and countries of the
European Economic Area), exported to third countries and
re-imported) and shall also carry out all the other tests and
checks that are necessary to ensure the quality of the medicinal
products in accordance with the requirements of the medicinal
product registration documentation. The quality control of
medicinal products shall not be carried out for the batches of
imported medicinal products that have undergone such controls in
another Member State of the European Community, and the medicinal
products have been supplied from another Member State together
with a control report signed by the qualified person.
15. The quality control of the medicinal products referred to
in Paragraph 14 of this Regulation need not be carried out if the
medicinal products are imported from the countries that have
entered into a mutual recognition agreement with the European
Community on the conformity assessment of good manufacturing
practice of medicinal products, and this agreement provides that
the testing of each batch of medicinal products (the qualitative
and quantitative analysis) is carried out in the exporting
country. In such case, the certificate of the batch of medicinal
products referred to in Paragraph 34 of this Regulation shall
accompany each batch of imported medicinal products.
[2 February 2016]
15.1 The exception referred to in Paragraph 15 of
this Regulation shall apply only to the manufacturing activities
or pharmaceutical forms that are indicated in the agreement
between the European Union and the respective country.
[2 February 2016]
16. The qualified person shall certify the batches of
medicinal products in all cases by making precise entries in a
registration logbook or in an equivalent document provided for
that purpose and by certifying with a signature that each batch
of the medicinal products has been manufactured and controlled in
conformity with the requirements referred to in Paragraphs 14 and
15 of this Regulation. The registration logbook or the equivalent
document shall be kept up to date as certain activities are
carried out and shall be kept at the undertaking at least for
five years after the making of the last entry, ensuring access to
such logbook or document to the officials of the State Agency of
Medicines and the Health Inspectorate.
[2 February 2016]
16.1 If the medicinal products are to be placed on
the market of the European Union, the qualified person shall
ensure that the packaging of the relevant medicinal products has
safety features referred to in Article 3(2)(a) and (b) of
Commission Delegated Regulation (EU) 2016/161 of 2 October 2015
supplementing Directive 2001/83/EC of the European Parliament and
of the Council by laying down detailed rules for the safety
features appearing on the packaging of medicinal products for
human use (hereinafter - Delegated Regulation No 2016/161).
[15 January 2019 / Paragraph shall be applied from 9
February 2019 in compliance with the transitional measures
specified in Articles 48 and 50 of Delegated Regulation No
2016/161. See Paragraph 56.2]
17. In order to carry out the quality control of the medicinal
products referred to in Paragraph 14 of this Regulation, a
quality control laboratory of another person may be used
(hereinafter - the contract acceptor), if the importer of
medicinal products and the contract acceptor enter into a written
contract in compliance with the requirements of Paragraphs 18, 19
and 20 of this Regulation.
18. The contract shall clearly define the obligations of the
parties, in particular, the obligation of the contract acceptor
to comply with the principles of and guidelines for good
manufacturing practice, and also the manner in which the
qualified person who is responsible for the certification of each
batch shall fulfil his or her obligations.
19. The contract acceptor shall ensure the fulfilment of the
following requirements:
19.1. if a written authorisation has not been received from
the importer of medicinal products, the contract acceptor shall
not enter into subcontract with a third person for the work which
has been entrusted to the contract acceptor in conformity with
the contract referred to in Paragraph 17 of this Regulation;
19.2. shall comply with the principles of and guidelines for
good manufacturing practice that have been specified in the laws
and regulations regarding the manufacture and control of
medicinal products and shall also subject oneself to the control
of the State Agency of Medicines.
20. Prior to entering into a contract for the performance of
the quality control of medicinal products, the importer of
medicinal products shall ensure that a statement is provided by
the State Agency of Medicines on the conformity of the laboratory
to the requirements for good manufacturing practice that have
been laid down in the guidelines of the European Commission for
good manufacturing practice for medicinal products and
investigational medicinal products. If the laboratory is located
in another country, the State Agency of Medicines shall establish
that the relevant laboratory has a valid certificate of good
manufacturing practice in the European Union database on
manufacturing and import licences and good manufacturing practice
certificates (EudraGMDP database) which includes quality control
of the relevant type of testing for which a contract is intended
to be entered into.
[14 July 2022]
21. In relation to the investigational medicinal products
which have been manufactured in third countries, the qualified
person shall be responsible for ensuring that each batch of the
medicinal products has been manufactured and checked in
conformity with the principles of and guidelines for good
manufacturing practice that are at specified in Chapter II of the
Commission Delegated Regulation No 2017/1569 and also in
conformity with the product specification and information
indicated by the sponsor in the submission to the State Agency of
Medicines for the receipt of the authorisation for the clinical
trial of medicinal products. For investigational medicinal
products from a third country used for comparison in a clinical
trial and registered in third countries but for which a
documentary evidence cannot be obtained that each series has been
produced under conditions that are at least equivalent to the
principles of good manufacturing practice laid down in Chapter II
of Commission Delegated Regulation No 2017/1569 and the
guidelines, the qualified person shall be responsible for the
analysis, testing and inspection of each batch of the preparation
in order to certify that the quality thereof conforms to the
information provided by the sponsor to the State Agency of
Medicines for the receipt of the authorisation for clinical
trials or an authorisation with a condition referred to in the
laws and regulations regarding clinical trials.
[14 July 2022]
22. In all cases, the qualified person shall make accurate
entries in the registration logbook or other equivalent document
intended for this purpose as regards the investigational
medicinal products and shall certify with a signature that each
batch of medicinal products conforms to the requirements referred
to in Paragraph 21 of this Regulation. After specified
activities, the registration logbook or the relevant document
shall be supplemented and stored in the undertaking for at least
five years from the completion or official termination of the
last study in which the specific batch of investigational
medicinal products was used and shall ensure access to the
aforementioned journal or document to officials of the State
Agency of Medicines.
[14 July 2022]
23. [14 July 2022]
24. The special authorisation (licence) for the
manufacture/importation of medicinal products referred to in
Paragraph 11 of this Regulation shall apply only to the medicinal
products (in relation to investigational medicinal products - to
the types of medicinal products and pharmaceutical forms) that
have been indicated by the importer of medicinal products in the
submission for the receipt of the special authorisation (licence)
for the manufacture/importation of medicinal products and that,
when issuing the aforementioned special authorisation (licence),
have been included by the State Agency of Medicines in the data
base in accordance with the laws and regulations regarding the
procedures for licensing the pharmaceutical activity.
[2 February 2016]
25. The importer of medicinal products may import narcotic and
psychotropic medicinal products that have been indicated in the
submission for the receipt of the special authorisation (licence)
for the manufacture or importation of medicinal products and
that, when issuing the aforementioned special authorisation
(licence), have been included by the State Agency of Medicines in
the database in accordance with the laws and regulations
regarding the procedures for licensing the pharmaceutical
activity if a single-use authorisation issued by the State Agency
of Medicines which corresponds to the requirements of the
Commission on Narcotic Drugs of the United Nations Organisation
(hereinafter - the UN) Economic and Social Council is issued for
the importation of the respective medicinal products in
accordance with the Law on the Legal Trade of Narcotic and
Psychotropic Substances and Medicinal Products, and also
Precursors. In addition to the requirements laid down in this
Regulation, the requirements specified in the Law on the Legal
Trade of Narcotic and Psychotropic Substances and Medicinal
Products, and also Precursors shall be complied with.
[14 July 2022]
26. The importer of medicinal products may import
non-prescription medicinal products that have been included in
the Register of Medicinal Products of the Republic of Latvia or
that have been centrally registered in accordance with Regulation
No 726/2004 of the European Parliament and of the Council on
behalf of budget institutions or public benefit
organisations.
[14 July 2022]
III. Importation of the Samples of
Medicinal Products and Unregistered Medicinal Products from Third
Countries
27. Unregistered medicinal products may be imported from third
countries by a person who, in accordance with the laws and
regulations regarding the licensing of pharmaceutical activity,
has received the special authorisation (licence) issued by the
State Agency of Medicines for the wholesale distribution of
medicinal products for human use and who has the authorisation
for the distribution of unregistered medicinal products issued by
the State Agency of Medicines in accordance with the procedures
specified in the laws and regulations regarding the distribution
and quality control of medicinal products for the individually
assigned medicinal products.
[14 July 2022]
27.1 Unregistered narcotic and psychotropic
medicinal products may be imported from third countries by a
person, if, in addition to the requirements referred to in
Paragraph 27 of this Regulation, a single-use authorisation
issued by the State Agency of Medicines which corresponds to the
requirements of the Commission on Narcotic Drugs of the UN
Economic and Social Council is issued for the import of the
specific medicinal products in accordance with the Law on the
Legal Trade of Narcotic and Psychotropic Substances and Medicinal
Products, and also Precursors.
[14 July 2022]
27.2 The requirements specified in Paragraph 27 of
this Regulation shall also apply to unregistered non-prescription
medicinal products referred to in Paragraph 26 of this
Regulation.
[14 July 2022]
28. Samples of medicinal products may be imported on the basis
of the authorisation referred to in Paragraph 28.1 of
this Regulation, if they are intended:
28.1. for the registration of medicinal products by submitting
them to the State Agency of Medicines;
28.2. for use in research or development testing in Latvia or
other countries (not applicable to investigational medicinal
products, including medicinal products used for comparison,
including placebo);
28.3. to be used for educational purposes;
28.4. to be used as samples of reference standards in
testing.
[14 July 2022]
28.1 Samples of medicinal products (except for the
samples of narcotic and psychotropic medicinal products) may be
imported from third countries by a person who has the
authorisation for the importation of the samples of medicinal
products into the Republic of Latvia issued by the State Agency
of Medicines (Annex 1). After the importation of the number of
packaging units of medicinal products indicated in the
authorisation, a new authorisation shall be needed for a
recurrent importation of medicinal products.
[2 February 2016]
28.2 Samples of narcotic drugs and psychotropic
medicinal products may be imported by a person who, in accordance
with the Law on the Legal Trade of Narcotic and Psychotropic
Substances and Medicinal Products, and also Precursors, has a
single-use authorisation issued by the State Agency of Medicines
which corresponds to the requirements of the Commission on
Narcotic Drugs of the UN Economic and Social Council. In this
case, the authorisation issued by the State Agency of Medicines
referred to in Paragraph 28.1 of this Regulation for
the importation of samples of medicinal products into the
Republic of Latvia shall not be necessary.
[14 July 2022]
29. In order to receive the authorisation for the importation
of the samples of medicinal products, the applicant for the
authorisation shall submit a submission to the State Agency of
Medicines in conformity with the requirements specified in Annex
2 to this Regulation. The submission shall include a
certification in which the need for importing samples of
medicinal products for the specific purpose specified in the
submission is justified.
[14 July 2022]
29.1 In order to receive the authorisation for the
importation of the samples of medicinal products, the following
persons shall be entitled to submit the submission:
29.1 1. a person who, in accordance with the laws
and regulations regarding the procedures for licensing
pharmaceutical activity, has received the special authorisation
(licence) issued by the State Agency of Medicines for the
manufacture or importation of medicinal products or the special
authorisation (licence) for the wholesale distribution of
medicinal products for human use;
29.1 2. the person referred to in Section
25.1 of the Pharmaceutical Law to whom the special
authorisation (licence) which gives the right to the wholesale or
manufacture of medicinal products has been issued in a Member
State of the European Union or a country of the European Economic
Area.
[14 July 2022]
29.2 The authorisation for the importation of the
samples of medicinal products shall indicate accordingly to whom
the holder of the authorisation is entitled to distribute the
samples of medicinal products:
29.2 1. samples for the registration of medicinal
products - to the person who submits medicinal products for
registration to the State Agency of Medicines;
29.2 2. samples for research or development testing
- to the person engaged in research or development testing in
Latvia or other countries;
29.2 3. samples for educational purposes - to an
educational institution;
29.24. for testing - a test laboratory in Latvia or
other countries.
[14 July 2022]
30. The State Agency of Medicines shall, within five working
days after receipt of the submission referred to in Paragraph 29
of this Regulation, verify whether the provided information meets
the requirements laid down in this Regulation. If the provided
information is incomplete or incorrect, the State Agency of
Medicines shall request in writing additional information.
31. The State Agency of Medicines shall take the decision to
refuse to issue the authorisation for the importation of the
samples of medicinal products, if the information requested
(justification) has not been received from the submitter of the
submission within one month after requesting the additional
information referred to in Paragraph 29 of this Regulation.
32. [14 July 2022]
32.1 The expenditures which are related to the
issue of the authorisation for the importation of the samples of
medicinal products shall be covered by the submitter of the
submission according to the price list of the paid services
provided by the State Agency of Medicines. The State Agency of
Medicines shall issue the authorisation for the importation of
the samples of medicinal products to the applicant for the
authorisation in the form of an electronic document within three
working days after taking the decision by sending it to the
electronic mail address indicated in the submission. The
authorisation in the form of a printed document shall be issued
upon a request within three working days for an additional fee
according to the price list of the paid services provided by the
State Agency of Medicines.
[14 July 2022]
IV. Exportation of Medicinal
Products
33. Medicinal products, including investigational medicinal
products and samples of medicinal products, may be exported by a
person who, in accordance with the laws and regulations regarding
the procedures for licensing pharmaceutical activity, has
received the special authorisation (licence) issued by the State
Agency of Medicines for the manufacture or importation of
medicinal products or the special authorisation (licence) for the
wholesale distribution of medicinal products for human use in
which a condition - the exportation of medicinal products - has
been specified (special authorisation (licence) for the opening
(operation) of a medicinal product wholesaler). The special
authorisation (licence) for the manufacture/importation of
medicinal products issued by the State Agency of Medicines shall
not be necessary for the freight of medicinal products that is
exported to third countries on the basis of the authorisation for
the manufacture/importation of medicinal products issued by a
competent authority of another European Union Member State, and
that is transported in transit through the territory of Latvia
(including placed in the customs warehouse).
[14 July 2022]
33.1 Narcotic and psychotropic medicinal products
and samples of medicinal products may be exported by a person
who, in addition to the special authorisations (licences)
referred to in Paragraph 33 of the Regulation, has a single-use
authorisation issued by the State Agency of Medicines that
corresponds to the requirements of the Commission on Narcotic
Drugs of the UN Economic and Social Council for the exportation
of the specific medicinal products in accordance with the
procedures specified by the Law on the Legal Trade of Narcotic
and Psychotropic Substances and Medicinal Products, and also
Precursors.
[14 July 2022]
33.2 The person who exports medicinal products
shall ensure the following:
33.2 1. the requirements specified in the
guidelines for good distribution practice of medicinal products
which are published by the European Commission (available in the
official language on the website of the State Agency of
Medicines) are complied with;
33.2 2. the medicinal products are supplied to such
persons in the third countries who are entitled to receive them
for the wholesale distribution or delivery thereof to population
in the third countries. For all supplies of medicinal products, a
document shall be enclosed where the following is indicated:
33.2 2.1. the date of supply;
33.2 2.2. the name of the medicinal product, the
form and strength or concentration of the medicinal product;
33.2 2.3. the supplied quantity (for each medicinal
product);
33.2 2.4. the name and address of the consignee and
supplier;
33.2 2.5. the manufacturing serial number of each
supplied batch of medicinal products.
[2 February 2016]
33.3 A person who has received the samples of
medicinal products referred to in Paragraph 28 of the Regulation
to be used for research or development testing, or as samples of
reference standards in testing, but the use of these samples for
research or development testing, or in testing is intended in a
third country may export such samples to the relevant country for
distribution to the recipient of the samples of medicinal
products in the third country indicated in the authorisation.
[14 July 2022]
34. If a manufacturer registered in Latvia exports medicinal
products to a country which has entered into a mutual recognition
agreement for the conformity assessment of medicinal product good
manufacturing practice with the European Community, a certificate
of the batch of medicinal products signed by the qualified person
in which information has been indicated in accordance with Annex
3 to this Regulation shall be attached to each batch of the
medicinal products to be exported.
35. The State Agency of Medicines shall, on the basis of a
submission of the medicinal product manufacturer, exporting
country, importing country or the competent authority of the
importing country shall issue the following:
35.1. a product certificate (Annex 4). A product, within the
meaning of this Paragraph, shall be such medicinal products in
the final pharmaceutical form thereof intended for humans and the
active substances for the use in such pharmaceutical forms,
which, in accordance with the procedures laid down in the laws
and regulations governing pharmaceutical activities, have been
subject to control in the exporting country and in the importing
country. The product certificate shall conform to the form
recommended by the World Health Organisation (WHO) and shall
determine the status of the product, as well as the status of the
certificate requester in the exporting country. The certificate
shall be intended only for a product of one type;
35.2. a statement on the licensing status of the
pharmaceutical product (Annex 5). This statement shall be
intended for a representative of the importer of medicinal
products who participates in international offers (tenders) in
accordance with the requirements of the invitation. This
statement shall denote that the specific medicinal products have
been registered in the Republic of Latvia (exporting country) and
they are permitted to be distributed. Upon request of the
applicant and the medicinal product registration owner, if they
are different persons, the State Agency of Medicines shall issue
the product certificate referred to in Sub-paragraph 35.1 of this
Regulation for each product referred to in the statement.
[13 August 2013]
35.1 The State Agency of Medicines shall, on the
basis of a submission by the manufacturer of medicinal products
or active substance (product) registered in Latvia or a request
of the competent authority of the third country, issue a product
certificate in an abridged format - a pharmaceutical product
certificate or free trade certificate (hereinafter - the abridged
certificate). The abridged certificate shall be issued per one
product type (in Latvian and English). If one medicinal product
has different strengths, they shall be considered as one type
product. If one medicinal product has different pharmaceutical
forms (for example, pellets, solution), a separate submission
shall be submitted and the abridged certificate shall be issued
for each pharmaceutical form.
[29 September 2009]
35.2 The abridged certificate shall be drawn up,
taking into account the requirements of the third country
specified in the submission insofar as they are not in
contradiction with the guidelines of the World Health
Organisation (WHO) for the certification scheme on the quality of
pharmaceutical products moving in international commerce. The
abridged certificate shall provide at least the following
information:
35.2 1. the certificate name: "Pharmaceutical
Product Certificate" or "Free Trade Certificate" in conformity
with the requirements of the third country;
35.2 2. the product name. Strength (quantity of
active substance(-s)
per strength, volume and mass unit) and pharmaceutical form
shall be specified for medicinal products. The international
non-proprietary name (INN) shall be specified for the active
substance, or, if there is not any, the chemical name. The
registration number, registration date and term of validity, if
any has been determined, shall be determined for the medicinal
products registered in Latvia;
35.2 3. the composition of the product. Strength
per one strength unit (packaging unit) shall be specified for the
active substance;
35.2 4. the name of the manufacturer (the firm name
of the merchant), unified registration number in the Commercial
Register, legal address, name, number, issuer, date of issue,
term of validity of the special authorisation (licence) (if any)
and address of the production unit;
35.2 5. the given name, surname or firm name and
legal address of the person responsible for placing on the market
of the medicinal product (applies to the medicinal products
registered in Latvia);
35.2 6. the given name, surname or firm name and
legal address of the person in whose name it is intended to
register medicinal products (applies to the medicinal products
submitted for registration in Latvia or in another country);
35.2 7. if medicinal products are under
registration procedure, it shall be specified. If the
registration of medicinal products is not intended, "not intended
to be registered for placing on the market in Latvia" or
"intended only for exportation" shall be indicated. If the
medicinal products or active substance may not be distributed in
Latvia, the relevant reason shall be specified, for example,
"registration suspended", "registration annulled" or
"registration refused". "Not to be registered" or "intended only
for exportation" shall be indicated for the active substance;
35.2 8. a certification that the relevant product
may be freely sold on the market of the particular third country
in accordance with the requirements laid down in the laws and
regulations of the relevant country and on the basis of quality
specifications of the particular manufacturer (specify the name)
and that the particular manufacturer subject to regular
inspection and certification procedure of good manufacturing
practice (specify the frequency of the carrying out of
inspections in accordance with the procedures laid down in the
laws and regulations regarding manufacture and control of
medicinal products) is responsible for the quality of the
aforementioned product.
[29 September 2009]
35.3 It shall be determined in the abridged
certificate that the quality specification of the compliant
product has been drawn up on the basis of the quality indicators
of the European Pharmacopoeia and the quality specification of
the manufacturer of the active substance, if the requester of the
abridged certificate has stipulated the necessity of such
information in a submission justifying it with the requirements
of the third country. This information need not be indicated if
the active substance has a certificate of conformity issued by
the European Directorate for the Quality of Medicines.
[14 July 2022]
35.4 A copy of the description of the medicinal
product, instructions for use and labelling shall be appended to
the abridged certificate. The abovementioned requirement shall
not apply to the abridged certificate form which is issued for an
active substance.
[29 September 2009]
36. In order to receive the product certificate referred to in
Sub-paragraph 35.1 of this Regulation, the medicinal product
manufacturer shall submit a submission to the State Agency of
Medicines in which the following shall be indicated:
36.1. the given name, surname or firm name and address, as
well as contact information (telephone, fax and electronic mail
address) of the applicant for the certificate;
36.2. the status of the applicant for the certificate:
36.2.1. manufactures the dosage form;
36.2.2. packages and labels the dosage form which is
manufactured by another independent manufacturer;
36.2.3. is not involved in the activities referred to in
Sub-paragraphs 36.2.1 and 36.2.2 of this Regulation;
36.3. if the applicant for a certificate is not the
manufacturer of the dosage form, the firm name and address of the
manufacturer of the dosage form shall be indicated;
36.4. the name, strength and dosage form of the product:
36.4.1. in Latvia;
36.4.2. in other countries;
36.5. the name of the active substances (by using the
international non-proprietary name (INNs) or national
non-proprietary name) and the amount thereof in one strength;
36.6. a full composition, including excipients (the
quantitative composition shall also be indicated if a consent
with the product registration certificate owner has been
attached);
36.7. whether the product has been registered in Latvia;
36.8. whether the product is distributed in Latvia;
36.9. the number of the product registration certificate and
date of issue (if necessary, whether the registration certificate
is provisional, or the product has not yet been approved shall be
indicated);
36.10. the given name and address of the medicinal product
registration owner;
36.11. the status of the medicinal product registration owner
in accordance with Sub-paragraphs 36.2.1, 36.2.2 and 36.2.3 of
this Regulation;
36.12. if the medicinal product registration owner is not the
manufacturer of the pharmaceutical form, the firm name and
address of the manufacturer of the pharmaceutical form shall be
indicated and a document certifying that the medicinal product
registration owner agrees to make such information available to
the public shall be attached;
36.13. if the registration certificate is not requested for
the product, one of the following reasons why it is not necessary
shall be indicated:
36.13.1. the product has been created only for special medical
treatment, mostly for the treatment of tropical diseases which
are not endemic in Latvia;
36.13.2. the product has been reformulated to improve its
stability under tropical conditions;
36.13.3. the product has been reformulated to exclude
excipients in the composition thereof not approved for use in the
importing country;
36.13.4. the product has been reformulated in order to create
another maximum possible strength limit for an active
substance;
36.13.5. other reasons (specify which);
36.14. if the status of the medicinal product registration
owner or the applicant for the certificate corresponds to the
status referred to in Sub-paragraph 36.2.2 or 36.2.3 of this
Regulation (especially if a foreign manufacturer is involved in
the manufacture of the product), the applicant for the
certificate shall submit information to the State Agency of
Medicines in which the conformity of each party involved in the
manufacture in relation to each stage of the manufacturing
process and the final product are determined, as well as the type
and amount of the control carried out by each party.
[13 August 2013]
37. In order to receive the statement on the licensing status
of pharmaceutical product referred to in Sub-paragraph 35.2 of
this Regulation, a person shall submit a submission to the State
Agency of Medicines in which the following is indicated:
37.1. the given name, surname or firm and address, as well as
contact information (telephone, fax and electronic mail address)
of the applicant for the certificate;
37.2. the importing country;
37.3. the name of the product, strength and pharmaceutical
form, the name of the active substances (by using the
international non-proprietary names (INNs) or national
non-proprietary names) and the amount thereof in one strength,
the number of the registration certificate and date of issue. If
the product has not been registered, "not required" or "not
requested", or "under consideration", or "refused" shall be
indicated as appropriate.
[13 August 2013]
37.1 In order to receive the abridged certificate
referred to in Paragraph 35.1 of this Regulation, a
medicinal product manufacturer registered in Latvia shall submit
a submission to the State Agency of Medicines. The submission
shall indicate:
37.1 1. the given name, surname or firm name and
address, as well as contact information (telephone, fax and
electronic mail address) of the applicant for the
certificate;
37.1 2. the third country, the competent
institution and the requirements to be specified in the abridged
certificate, as well as the requirements of the third country for
the validity of the certificate of good manufacturing practice,
if any have been determined;
37.1 3. the information referred to in
Sub-paragraphs 35.2 1, 35.2 2,
35.2 3, 35.2 4, 35.2 5,
35.2 6 and 35.1 7 of this Regulation. If
medicinal products have been submitted for registration, the
country (countries) where the submission for registration has
been submitted shall be specified. If the product has not been
registered in Latvia, the country (countries)
in which the product has been registered shall be
specified;
37.1 4. if the information referred to in Paragraph
35.3 of this Regulation is to be included in the
abridged certificate, the quality specification of the product
shall be appended to the submission. The quality specification
(specifications) of the manufacturer of active substance
(substances) shall also be submitted for the medicinal
products.
[29 September 2009; 13 August 2013]
38. The State Agency of Medicines shall issue the product
certificate and the statement on the licensing status of the
pharmaceutical product referred to in Paragraph 35 of this
Regulation within 30 days after receipt of the submission. The
expenditures associated with the issue of the product certificate
shall be covered by the submitter of the submission in accordance
with the price list of the paid services provided by the State
Agency of Medicines. The State Agency of Medicines shall issue
the certificate and the statement on the licensing status of the
pharmaceutical product in the form of an electronic document,
sending it to the electronic mail address indicated in the
submission. The certificate and statement or its duplicate shall,
upon request, be issued in the form of a printed document within
three working days for an additional fee in accordance with the
price list of the paid services provided by the State Agency of
Medicines.
[13 August 2013; 2 February 2016]
38.1 The State Agency of Medicines shall issue the
abridged certificate referred to in Paragraph 35.1 of
this Regulation within 30 days after receipt of a submission. If
the product certificate referred to in Sub-paragraph 35.1 of this
Regulation has been issued for the product and repeat assessment
of good manufacturing practice or assessment of the data referred
to in Paragraph 35.3 of this Regulation is not
necessary, the State Agency of Medicines shall issue the abridged
certificate referred to in Paragraph 35.1 of this
Regulation within 10 days after receipt of the submission. The
expenditures associated with the issue of the abridged
certificate shall be covered by the submitter of the submission
in accordance with the price list of the paid services of the
State Agency of Medicines.
[29 September 2009; 2 February 2016]
38.2 The State Agency of Medicines shall issue the
abridged certificate in the form of an electronic document,
sending it to the electronic mail address indicated in the
submission. The certificate and statement or its duplicate shall,
upon request, be issued in the form of a printed document within
three working days for an additional fee in accordance with the
price list of the paid services provided by the State Agency of
Medicines.
[13 August 2013; 2 February 2016]
38.3 The submission for the issue of the product
certificate referred to in Sub-paragraph 35.1 of this Regulation,
the statement on the product licensing status referred to in
Sub-paragraph 35.2 of this Regulation and the abridged
certificate referred to in Paragraph 35.1 of this
Regulation may be submitted in the form of an electronic
document, preparing it in accordance with the laws and
regulations regarding drawing up of electronic documents.
[13 August 2013]
39. If an inspection of the manufacturing site of the active
substance and conformity assessment of good manufacturing
practice of the active substance is necessary for the issue of
the certificate referred to in Sub-paragraph 35.1 of this
Regulation for a product that is an active substance to be used
in pharmaceutical form, the applicant for the certificate shall
request the State Agency of Medicines to carry out the conformity
assessment of the manufacturing of the active substances. The
abovementioned inspection shall be carried out and the
certificate of good manufacturing practice shall be issued by the
State Agency of Medicines in accordance with the procedures laid
down in the laws and regulations regarding the manufacture and
control of medicinal products.
[10 June 2008]
39.1 Should an inspection of the manufacturing site
of the product and conformity assessment of good manufacturing
practice need to be made for issuing the abridged certificate
referred to in Paragraph 35.1 of this Regulation, the
applicant for the certificate shall request the State Agency of
Medicines to carry out the conformity assessment of good
manufacturing practice and to issue the certificate of good
manufacturing practice. Such inspection shall be carried out and
the certificate of good manufacturing practice shall be issued by
the State Agency of Medicines in accordance with the procedures
laid down in the laws and regulations regarding the manufacture
and control of medicinal products.
[29 September 2009]
V. Supervision
and Sanctions
40. The Food and Veterinary Service shall:
40.1. control the conformity of the importation of medicinal
products with the requirements specified in Sub-paragraphs 8.3
and 9.1 of this Regulation and in Paragraphs 6, 11, 25, 27,
27.1, 27.2, 28, 28.1,
28.2, 43 and 43.1 of this Regulation;
40.2. control the importation of medicinal products and also
the conformity of transport and storage conditions (including
temperature regime) with the instructions of the medicinal
product manufacturer;
40.3. provide information to the Health Inspectorate on the
violations of the requirements laid down in this Regulation.
[10 June 2008; 29 September 2009; 14 September 2010; 2
February 2016; 14 July 2022]
41. The Food and Veterinary Service is entitled to prohibit
the importation of medicinal products:
41.1. in the following cases:
41.1.1. the accompanying freight documents do not conform to
the requirements laid down in Sub-paragraph 9.1 of the Regulation
or the medicinal products cannot be identified (there is no
labelling);
41.1.2. the imported medicinal products are not indicated in
the database of the State Agency of Medicines in accordance with
the laws and regulations regarding the procedures for licensing
pharmaceutical activity;
41.1.3. the medicinal products do not have the relevant
licences or authorisations referred to in Paragraphs 11, 25, 27,
27.1, 27.2, 28, 28.1, and
28.2 of the Regulation or they are not valid;
41.1.4. the requirements for the transportation of medicinal
products (including the storage temperature regime) have been
violated;
41.1.5. the medicinal products have expired;
41.1.6. the consignor and the consignee of the freight of the
medicinal products cannot be identified;
41.2. in accordance with Article 9 of the Council Regulation
No 2016/793, if it has been determined that the imported
medicinal products are the tiered-priced products that have been
included in Annex 1 to the Council Regulation No 2016/793;
41.3. in accordance with Article 14 of the Council Regulation
No 816/2006, if there are grounds for suspecting that the
importation prohibition in relation to the medicinal products
that have been manufactured in accordance with a compulsory
licence laid down in Article 13(1) of the Council Regulation No
816/2006 has been breached;
41.4. if the special authorisation (licence) for
pharmaceutical activity issued to a merchant who imports the
medicinal products referred to in Sub-paragraph 41.1.2 of this
Regulation is not in effect;
41.5. if the rapid alert statement of the Health Inspectorate
on a suspected quality defect and withdrawal of medicinal
products from the market applies to the imported medicinal
products in accordance with the laws and regulations regarding
the procedures for the distribution and quality control of
medicinal products;
41.6. if there are suspicions of possibly falsified medicinal
products.
[14 July 2022]
42. The Food and Veterinary Service shall notify the Health
Inspectorate of the decision on the day when it is taken.
[14 July 2022]
43. [1 January 2023 / See Paragraph 56.5]
43.1 Medicinal products the importation of which is
prohibited in accordance with Paragraph 41 of this Regulation
shall be diverted to the customs warehouse which has received the
special authorisation (licence) referred to in Paragraph
7.1 of this Regulation.
[14 July 2022 / Paragraph shall come into force on 1
January 2023. See Paragraph 56.6]
44. If the Food and Veterinary Service has prohibited the
importation of medicinal products in accordance with Paragraph 41
of the Regulation, the Health Inspectorate shall, after final
clarification of the circumstances, take the decision to revoke
the suspension of the importation of medicinal products or on the
further movement of the medicinal product and shall notify the
customs authority of the decision taken.
[14 July 2022]
45. [2 February 2016]
46. The expenditures associated with the disposal or
re-exportation of the specific freight of medicinal products
shall be covered by the person to whom the prohibition of placing
medicinal products on the market (the possessor of the medicinal
products) provided for in Regulation No 765/2008 of the European
Parliament and of the Council refers.
[2 February 2016]
47. The State Agency of Medicines shall control the conformity
of the importer of medicinal products with the requirements laid
down in Paragraphs 12, 14, 15, 16, 17, 18, 19, 20, 21, 22 and 23
of this Regulation in accordance with the procedures laid down in
laws and regulations regarding the manufacture and control of
medicinal products.
48. The importer of medicinal products shall provide the
following data to the officials of the State Agency of Medicines
during the check:
48.1. data on the quality control of each batch of the
medicinal products (that has been carried out in a country of the
European Economic Area) in accordance with the medicinal product
registration documentation;
48.2. all copies of the control reports approved by a
qualified person on immunological preparations and medicinal
products derived from human blood or plasma.
49. The Health Inspectorate shall, based on a report of the
State Agency of Medicines, be entitled to suspend the importation
of the medicinal products referred to in a file of the special
authorisation (licence) of the importer for specific or all
medicinal products or, by evaluating each case separately in
cooperation with the State Agency of Medicines, to decide on
suspension of the importation of medicinal products if:
49.1. the quality control of the medicinal products and the
batch release do not conform to the requirements laid down in
Paragraphs 14, 15 and 16 of this Regulation;
49.2. the qualified person does not fulfil the obligations
laid down in Paragraphs 14, 15 and 16 of this Regulation (in
relation to the investigational medicinal products - Paragraphs
21 and 22 of this Regulation);
49.3. the importer of medicinal products does not provide the
data and information laid down in Paragraph 48 of this Regulation
during the check;
49.4. the importer of medicinal products performs its
pharmaceutical activities in the area (address) and premises that
are not indicated in the respective special authorisation
(licence) and submission for the receipt thereof, as well as in
the file of the licence;
49.5. the importer of medicinal products imports the medicinal
products that are not indicated in the respective submission for
the receipt of the special authorisation (licence) and file of
the licence (this shall not refer to samples of medicinal
products and unregistered medicinal products);
49.6. the importer of medicinal products has no qualified
personnel whose qualification and professional experience
correspond to the requirements laid down in the laws and
regulations regarding the procedures for the manufacture and
control of medicinal products;
49.7. manufacture of the imported medicinal products or active
substances of medicinal products does not comply with the
requirements for good manufacturing practice of medicinal
products or active substances;
49.8. medicinal products or active substances of medicinal
products are falsified.
[10 June 2008; 2 February 2016]
50. The State Agency of Medicines shall fulfil the obligations
of the competent supervisory authority referred to in Article 19
of Regulation No 726/2004 of the European Parliament and of the
Council in relation to medicinal products that in accordance with
the aforementioned Regulation have been registered under the
centralised licensing procedure and have been imported from third
countries.
51. The Health Inspectorate shall:
51.1. monitor whether the distribution and transportation of
medicinal products in the areas referred to in Paragraph
2.1 of this Regulation complies with the requirements
laid down in this Regulation, laws and regulations regarding the
distribution of medicinal products, and the guidelines for good
distribution practice of medicinal products published by the
European Commission (available in the official language on the
website of the State Agency of Medicines);
51.2. [1 January 2023 / See Paragraph 56.7];
51.3. is entitled to request and receive information which is
related to the fulfilment of this Regulation from the State
Agency of Medicines, the Food and Veterinary Service and other
competent State institutions;
51.4. provide the necessary information to the State Agency of
Medicines, the Food and Veterinary Service and other competent
State institutions;
51.5. inform the European Commission of all decisions that
have been taken in accordance with the fulfilment of the
requirements of Council Regulation No 2016/793 of the European
Parliament and of the Council;
51.6. inform the European Commission of any decisions in
relation to the confiscation or disposal of products that have
been taken in accordance with Regulation No 816/2006 of the
European Parliament and of the Council.
[10 June 2008; 29 September 2009; 14 September 2010; 2
February 2016; 14 July 2022 / See Paragraph
56.7]
52. The officials of the relevant institutions shall not
disclose trade secrets of the controlled person which have become
known to them during the performance of their official duties in
accordance with this Regulation.
53. The Health Inspectorate, the State Agency of Medicines,
the Food and Veterinary Service and the customs authorities
shall, within the scope of their competence, ensure prompt mutual
exchange of information, as well as, in order to prevent the
diversion of medicinal products to illegal circulation, provide
information to the law enforcement institutions and the Ministry
of Health on the facts of which they have become aware.
[10 June 2008; 29 September 2009]
53.1 The Health Inspectorate, the State Agency of
Medicines and the Food and Veterinary Service shall co-operate
within the scope of their competence to ensure that the medicinal
products which are imported and which are not intended to be
placed on the market of the European Union do not enter
circulation, if there are justified suspicions that they are
falsified.
[13 August 2013]
53.2 The customs authorities shall notify the
Health Inspectorate and the State Agency of Medicines without
delay, if there are suspicions of possibly falsified medicinal
products.
[13 August 2013]
VI. Closing Provisions
54. Cabinet Regulation No. 88 of 27 February 2001, Regulations
regarding the Import, Export and Distribution of Medicinal
Products and Requirements for the Opening and Operation of
Medicinal Product Wholesalers (Latvijas Vēstnesis, 2001,
No. 35, 52; 2003, No. 114; 2004, No. 69), is repealed.
55. The medicinal product wholesalers to which, on the day of
coming into force of this Regulation, the special authorisation
(licence) for the opening (operation) of a medicinal product
wholesaler with a condition of special activity - the importation
of medicinal products into Latvia from a country which is not
located in the European Economic Area, and the authorisation
issued by the State Agency of Medicines for the importation of
the respective medicinal products into the Republic of Latvia
from third countries has been issued are entitled to import
medicinal products until the receipt of the special authorisation
(licence) for the manufacture/importation of medicinal products
referred to in the laws and regulations laying down the
procedures for the issuing, suspension, re-registration, and
revocation of the special authorisation (licence) for
pharmaceutical activity, but not longer than until 1 January
2008.
56. The sponsor to which, on the day of coming into force of
this Regulation, the authorisation for the import of medicinal
products for human use intended for clinical trials into the
Republic of Latvia has been issued by the State Agency of
Medicines is entitled to import the investigational medicinal
products until the receipt of the special authorisation (licence)
for the manufacture/importation of the medicinal products
referred to in the laws and regulations laying down the
procedures for the issuing, suspension, re-registration, and
revocation of the special authorisation (licence) for
pharmaceutical activity, but not longer than until 1 January
2008.
56.1 The requirement which applies to the issuing
of the documents referred to in Paragraphs 32.1, 38,
38.2 and 38.3 of this Regulation in the
form of a printed document for an additional fee shall be
applicable from 1 July 2014.
[13 August 2013]
56.2 Paragraph 16.1 of this Regulation
shall be applied from 9 February 2019 in compliance with the
transitional measures specified in Articles 48 and 50 of
Delegated Regulation No 2016/161.
[15 January 2019]
56.3 Paragraph 7 of this Regulation shall be in
force until 31 December 2022.
[14 July 2022]
56.4 Paragraph 7.2 of this Regulation
shall come into force on 1 January 2023.
[14 July 2022]
56.5 Paragraph 43 of this Regulation shall be in
force until 31 December 2022.
[14 July 2022]
56.6 Paragraph 43.1 of this Regulation
shall come into force on 1 January 2023.
[14 July 2022]
56.7 Sub-paragraph 51.2 of this Regulation shall be
in force until 31 December 2022.
[14 July 2022]
57. This Regulation shall come into force on 1 August
2007.
Informative Reference to the
European Union Directives
[29 September 2009; 13 August
2013; 25 September 2018; 14 July 2022]
This Regulation contains legal norms arising from:
1) [25 September 2018 / See Paragraph 2 of Amendments];
2) Directive 2001/83/EC of the European Parliament and of the
Council of 6 November 2001 on the Community code relating to
medicinal products for human use;
3) [25 September 2018 / See Paragraph 2 of Amendments];
4) Directive 2004/27/EC of the European Parliament and of the
Council of 31 March 2004 amending Directive 2001/83/EC on the
Community code relating to medicinal products for human use;
5) [14 July 2022];
6) [25 September 2018 / See Paragraph 2 of Amendments];
7) Commission Directive (EU) 2017/1572 of 15 September 2017
supplementing Directive 2001/83/EC of the European Parliament and
of the Council as regards the principles and guidelines of good
manufacturing practice for medicinal products for human use;
8) Directive 2011/62/EU of the European Union and of the
Council of 8 June 2011 amending Directive 2001/83/EC on the
Community code relating to medicinal products for human use, as
regards the prevention of the entry into the legal supply chain
of falsified medicinal products.
Prime Minister A. Kalvītis
Acting for the Minister for Health,
Minister for Welfare D. Staķe
Annex 1
Cabinet
Regulation No. 436
26 June 2007
[14 July 2022]
Authorisation for the Importation
of the Samples of Medicinal Products into the Republic of
Latvia
STATE AGENCY OF
MEDICINES
|
| (legal address, telephone number, fax number,
electronic mail address) |
Rīga
|
On the basis of decision No. |
|
of the State Agency of Medicines |
|
| |
(date) |
|
|
on the issuing of the authorisation for the importation of the
samples of medicinal products from third countries
|
| (name, type, registration number of the legal
person) |
|
| (submission
for the receipt of authorisation No. |
|
), |
| |
(registration number with the State Agency of
Medicines, date of submission and registration) |
|
is allowed to import the following samples of medicinal
products in the Republic of Latvia from third countries:
| Name of the
sample of medicinal products, dosage form, strength, size of
packaging |
Number of the
samples of medicinal products |
Purpose of use
of the samples of medicinal products |
It is allowed
to receive the samples of medicinal products |
Medicinal
product manufacturer, country |
| 1 |
2 |
3 |
4 |
5 |
| |
|
|
|
|
| |
|
|
|
|
|
Director of the State Agency of Medicines |
|
| |
(given name, surname, signature) |
Place for a seal
Notes.
1. In a Table's column or row, which is not completed, draw a
dash.
2. In column 3 of the Table, the purpose of the use of the
samples of medicinal products - "Submission to the State Agency
of Medicines in relation to the registration of medicinal
products", "Use for scientific purposes", "Use for educational
purposes", or "Use in the testing of medicinal products as the
sample of reference standards" shall be indicated.
3. In column 4 of the Table, the person to whom the
authorisation owner is entitled to distribute the samples of
medicinal products referred to in the authorisation shall be
indicated by including an indication:
3.1. "To the applicant for the receipt of the medicinal
product registration certificate" and its name and country;
3.2. "To the medicinal products registration owner" and its
name and country;
3.3. "To the scientific research institution" and its name,
number of the taxpayer certificate of the State Revenue Service
in the State Revenue Service Value Added Tax Taxable Persons
Register;
3.4. "To the educational institution" and its name, number of
the taxpayer certificate of the State Revenue Service in the
State Revenue Service Value Added Tax Taxable Persons
Register;
3.5. "To the test laboratory" and its name, number of the
taxpayer certificate of the State Revenue Service in the State
Revenue Service Value Added Tax Taxable Persons Register.
4. The details of the document "signature" and "place for a
seal" shall not be completed if the electronic document has been
prepared in accordance with the laws and regulations regarding
drawing up of electronic documents.
Annex 2
Cabinet
Regulation No. 436
26 June 2007
[14 July 2022]
Submission for the Receipt of the
Authorisation for the Importation of the Samples of Medicinal
Products into the Republic of Latvia
Hereby we ask the State Agency of Medicines to issue the
authorisation for the importation of the samples of medicinal
products into the Republic of Latvia from third countries with
the aim (mark as appropriate with an X):
 to submit them to the State Agency of Medicines in
relation to the registration of medicinal products
to submit them to the State Agency of Medicines in
relation to the registration of medicinal products
to use them for research or development
testing
to export to another country
to use for educational purposes
to use them in the testing of medicinal products
as the sample of reference standards
to export to another country
We want to receive the authorisation in a printed form (mark
as appropriate with an X):
yes
no
Part I
Information on the applicant and medicinal product
|
1. Applicant:
number of the taxable person for value added tax in the
State Revenue Service Value Added Tax Taxable Persons
Register
|
|
| 1.3.
address of the place of operation |
|
|
|
|
|
| 1.4. telephone number |
|
fax number |
|
| |
|
|
|
| 1.5. official electronic
address (if the submitter of the submission does not
use the official electronic address - electronic mail
address) |
| |
|
| |
|
2. Samples of medicinal products:
|
|
| 2.2.
form of the medicinal product |
|
|
|
| 2.3.
active ingredient and strength or concentration |
|
|
|
2.5.
registration number in the Medicinal Product Register
of Latvia
(for registered medicinal products) |
|
|
|
| 2.6.
amount of packages (number) |
|
|
| 3. Consignor of the samples
of medicinal products, its address, telephone number,
fax number, electronic mail address |
| |
|
| |
|
| 4. Medicinal product manufacturer: |
| 4.1.
name |
|
|
|
| 4.2.
legal address and address of the workplace of the
undertaking |
|
|
|
|
|
| 4.3. telephone number |
|
fax number |
|
| |
|
|
|
| Person to contact in connection
with the submission (given name, surname, telephone
number, fax number, official electronic address (if the
submitter of the submission does not use the official
electronic address - electronic mail address)) |
| |
|
| |
|
|
Part II
Appended Documents
|
Appended
Documents
|
Mark as appropriate
with X, No. of pages |
| 1. A certification that the samples of
medicinal products have been received from the medicinal
product manufacturer (if the samples are imported for the
registration of medicinal products) or from a person who has
the right to distribute medicinal products in the exporting
country |
|
| 2. A certification that the samples of
medicinal products are intended for submission to the State
Agency of Medicines in relation to the registration of
medicinal products |
|
3. A certification that the samples of medicinal products
are intended to be used for research or development testing
|
|
|
|
| (name
of the study and address of the study/development test
location) |
|
|
4. A certification that the samples of medicinal products
are intended to be used for educational purposes
|
|
|
|
| (name,
location of the educational institution, speciality
(study programme)) |
|
|
5. A certification that the samples of medicinal products
are intended to be used as the samples of reference
standards in the testing of medicinal products
|
|
|
|
| (name
and address of the test laboratory) |
|
|
|
6. If the study or development testing of the samples of
medicinal products or the testing of the samples of
reference standards is intended in another country:
6.1. an authorisation or a certification issued by the
competent authority of the relevant country or the public
information of the competent authority (for example,
whether the merchant has a valid special authorisation
(licence) which allows to purchase the specific medicinal
products) that the recipient of the samples of medicinal
products indicated by the submitter is entitled to receive
the samples of medicinal products
|
|
6.2. the name of the recipient
|
|
|
|
|
(identification number in the enterprise register or
taxpayer register of the relevant country) |
|
|
|
6.3. address, number of the special authorisation (licence)
(if any), identifier of the study (if any)
|
|
| |
|
| I, |
|
| |
(given name, surname, position of the responsible
official (an authorised representative of the
applicant)) |
certify that the information provided by me is true.
Responsible official
(authorised representative of the applicant)
|
| (position, given name, surname, signature) |
|
|
Date |
|
| |
(date of receipt of the submission in the State
Agency of Medicines) |
Notes.
1. In a column or row, which is not completed, draw a
dash.
2. If the form is sent without using electronic data media,
the applicant shall sign each page appended to the form.
3. If the submission is drawn up on several pages, the
responsible official shall sign each page.
4. The detail of the document "signature" shall not be
completed if the electronic document has been prepared in
accordance with the laws and regulations regarding drawing up of
electronic documents.
Annex 3
Cabinet
Regulation No. 436
26 June 2007


Acting for the Minister for Health,
Minister for Welfare D. Staķe
Annex 4
Cabinet
Regulation No. 436
26 June 2007
[13 August 2013]

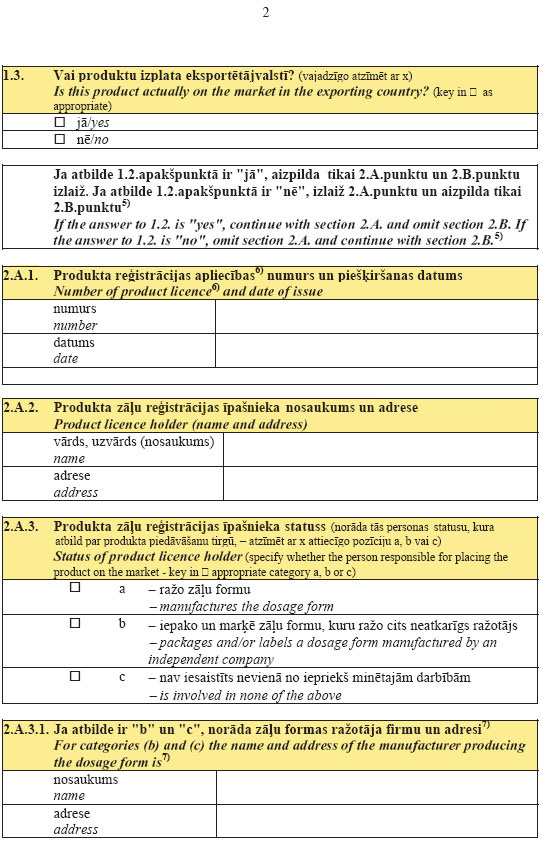
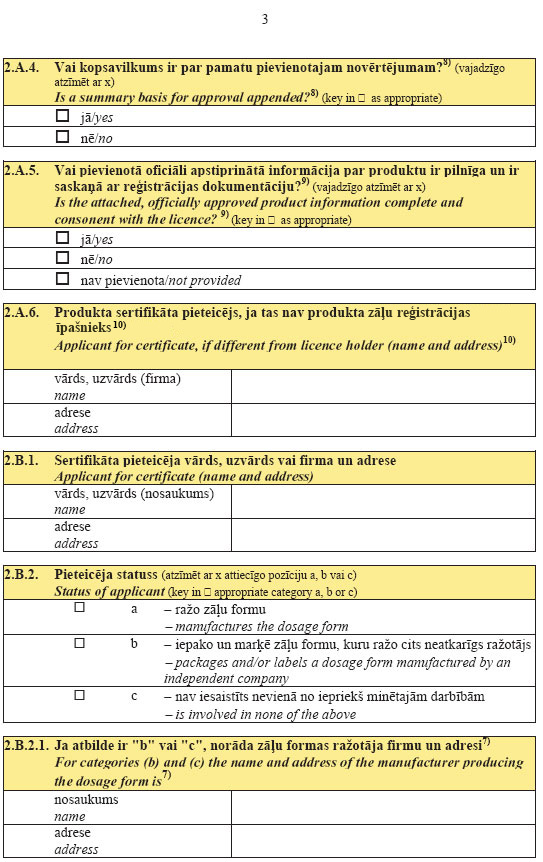
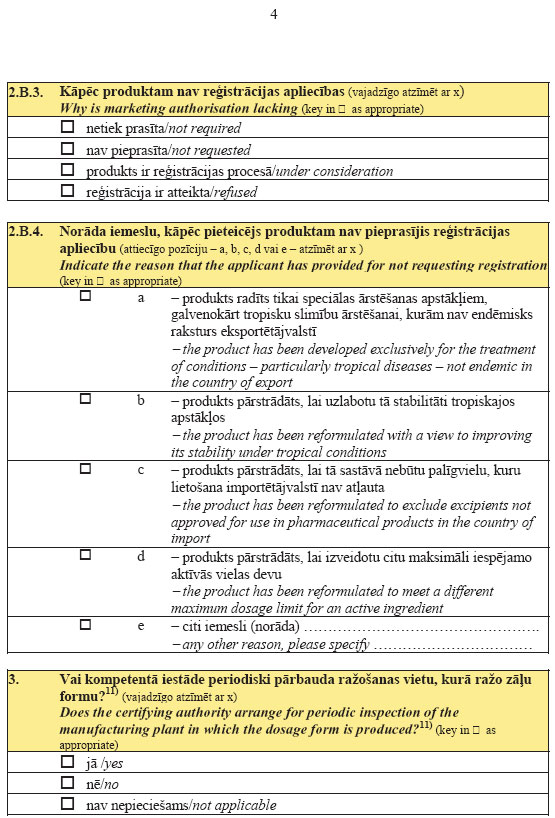



Acting for the Minister for Health,
Minister for Welfare D. Staķe
Annex 5
Cabinet
Regulation No. 436
26 June 2007
[13 August 2013]
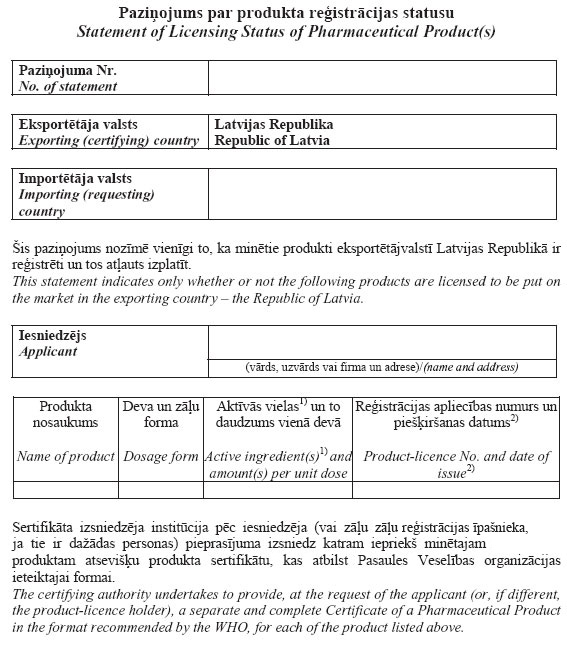

Acting for the Minister for Health,
Minister for Welfare D. Staķe
Translation © 2023 Valsts valodas centrs (State
Language Centre)








